Document Type : Original Article
Authors
1 Graduate Program in Bioclinical Sciences, Chulabhorn International College of Medicine, Thammasat University (Rangsit Campus), Pathumthani, 12120, Thailand
2 Center of Excellence in Pharmacology and Molecular Biology of Malaria and Cholangiocarcinoma, Thammasat University (Rangsit Campus), Pathumthani, 12120, Thailand
3 Drug Discovery and Development Center, Office of Advanced Science and Technology, Thammasat University (Rangsit Campus), Pathumthani, 12120, Thailand
Abstract
Cholangiocarcinoma remains a problem in several tropical and subtropical parts of the world, especially in Southeast Asia. The study determined plasma protein binding and blood-to-plasma ratio (B:P) of atractylodin (AT) and β-eudesmol (BE), the two active constituents of Atractylodes lancea (Thunb.) D.C., which is currently under development as an alternative treatment for cholangiocarcinoma. Plasma protein binding studies were performed using the ultracentrifugation method. Plasma samples (n=10) were spiked with AT or BE (2.5, 5, and 10 µg/mL) and incubated at 37 °C for 1 h. Blood to plasma (B:P) ratios of both compounds (0.5 mg/ml) were determined in human plasma produced from the spiked whole blood and the spiked plasma (n=10 each) and incubated at 37 °C for 10 min and 1 hour, respectively. Concentrations of AT and BE were determined using HPLC-UV. Plasma protein binding (%PPB) of AT and BE were relatively high (>90%), with low unbound fractions (fu < 0.05). The AT and BE concentrations in plasma and red blood cells (RBCs) were comparable, with the KRBC/Plasma (B:P) ratio of about 1. The high partitioning in blood circulation and high plasma protein binding of AT and BE may limit the extent of their distribution and delivery to the target tissues (bile ducts). Either plasma or whole blood can be used as the matrix of choice for pharmacokinetic studies. The information obtained would provide essential input parameters in the pharmacokinetic models to predict optimal dose regimens of AL in clinical studies.
Graphical Abstract
)
Keywords
- Antractylodes lancea (Thunb.) D.C. Atractylodin
- Eudesmol Plasma protein binding Blood
- to
- plasma ratio
Main Subjects
Introduction
The highest incidence and mortality rate of cholangiocarcinoma (CCA), a biliary tract cancer, CCA is reported in Asia, especially in the Northeastern region of Thailand [1]. Lack of effective chemotherapeutic drugs is a significant factor that limits effective CCA control. Atractylodes lancea (Thunb). D.C. (AL) is currently under development by our research team to control cholangiocarcinoma [2-4]. The crude extract of AL rhizomes has traditionally been used in China, Japan, and Thailand to treat various diseases and health conditions, including rheumatic diseases, digestive disorders, night blindness, influenza, and cancer [2-4]. Atractylodin (AT) and β-eudesmol (BE) are major bioactive constituents of AL which account for these pharmacological activities [5, 6]. The crude ethanol extract of AL rhizomes and the bioactive compounds AT and BE show promising anti-cholangiocarcinoma activity and safety profiles in a series of in vitro, in vivo, and clinical studies [7-12]. The capsule formulation of the standardized AL extract has been developed for clinical use in CCA recent patients [13]. The pharmacokinetic studies of AT and BE in animals and humans suggest rapid absorption, distribution, and excretion of both compounds [7-12]. These result in low and short systemic bioavailability after oral administration. Further dose optimization using pharmacokinetic modeling requires the input of key physicochemical parameters. The previous study [1] determined lipophilicity, aqueous solubility, and degree of AT and BE ionization. The LogP (partition coefficient) and LogD (distribution coefficient) values of 3.0-5.0 suggest moderate lipophilicity of both compounds. In addition, the aqueous solubility of AT (0.08-0.93) and BE (1.97-3.48) are low. Both are basidic compounds (pKa 9.63 and 9.12). These physicochemical properties of AT and BE may limit the bioavailability of AL following oral dose administration. The present study further determined plasma protein binding and blood-to-plasma ratios of AT and BE in vitro. Plasma protein binding and blood partitioning of drugs are known to influence their pharmacokinetics and clinical efficacy.
Evaluating both parameters of the candidate compounds would provide an essential piece of information during drug development. Information about the distribution of compounds in blood would further help to determine the suitable biological matrix for therapeutic drug monitoring and pharmacokinetic studies.
Materials and Methods
Chemicals and reagents
Atractylodin (AT) and β-eudesmol (BE were purchased from Wako Pure Chemical Industries (Osaka, Japan). Sigma Chemical (St. Louis, MO, USA) supplied dimethyl sulfoxide (DMSO) and other chemicals, including organic solvents. Ultrapure analytical grade Type I water (r > 18 MW/cm, Milli-Q PlusTM water system, Millipore Corporation, Bedford, MA, USA) was used in all experiments.
AT and BE stock solutions (in DMSO) were diluted with potassium phosphate buffer (0.1 M, pH 7.4) to the desired concentrations and stored at -20 °C. The final DMSO concentration was lower than 0.1%.
HPLC analysis
AT and BE concentrations in plasma samples (produced from spiked whole blood and spiked plasma) were determined by high-performance liquid chromatography (HPLC: Thermo Fisher Scientific, CA, USA) [8]. The system consisted of the elution solvent delivery (SpectraSystem P4000 Quaternary Solvent Delivery/Controller), solvent degasser (SpectraSystem SCM1000 Solvent Degasser), auto-sampler (SpectraSystem AS3500), the UV detector (SpectraSystem UV/Vis 3000, 340 nm), reversed-phase column (Thermo Hypersil Gold C18, 250 mm × 2.1 mm i.d., 5 μm), and elution solvent (70% acetonitrile: 30% distilled water, 1 mL/min). Aliquots of samples (200 μL) were injected onto the column.
Sample preparation
Various concentrations of AT or BE (20 μL) were added to plasma (1 mL), and thoroughly mixed. 2 mL of Acetonitrile (2 mL) was added, and then the mixture was vortexed (30 sec) and centrifuged (3,000 xg, 10 min). The supernatant was extracted with dichloromethane (4 mL) by rotatory mixing (30 min). The organic phase (upper layer) was separated through centrifugation (3,000xg, 10 min) and evaporated to dryness under the nitrogen stream (40 °C). An aliquot of a sample (40 μL) reconstituted with the mobile phase (100 μL) was injected into the column. The calibration curves of AT and BE were linear over the concentration range of 2.5-500 ng/mL (r > of 0.999). The accuracy varied between 0.2% and 6.1%, and the precision (coefficients of variation: CV) were below 5%. The analytical recovery for both AT and BE in plasma samples were 75.6-77.4%. The limit of quantification (LOQ) was 2.5 ng/mL using 1 mL plasma.
In vitro assay for plasma protein binding
Plasma protein binding of AT and BE was determined using an ultrafiltration method [11]. Outdated human plasma samples used in the experiment were obtained from the. AT or BE was spiked to plasma (1 mL, Thammasat Chalermprakiet Hospital, Pathumtanee, Thailand) to obtain final concentrations of 2.5, 5, and 10 µg/mL and equilibrated at 37 °C for 1 h. Unbound (free) AT or BE in plasma sample (1 mL) was separated from the protein-bound compounds using an ultrafiltration system with Centrifree® 30K MWCO membrane and centrifugation (30,000 rpm, 6 min), and the concentrations of free AT and BE in plasma filtrate were measured using HPLC, as described above. The assay was done three times.
Plasma protein binding of each compound is expressed as fraction unbound (fu) or % protein binding as follows:
Fraction unbound (fu)
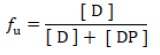
Plasma protein binding (% PPB)
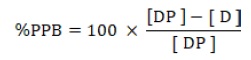
Where, [D] is the free drug concentration and [DP] is the concentration of the drug-protein complex.
In vitro assay for blood-to-plasma concentration ratios
AT or BE was spiked in plasma and whole blood samples (n=10,1 mL each, Thammasat Chalermprakiet Hospital, Pathumtanee, Thailand) to obtain 0.5 µg/ml final concentration. The sample was equilibrated (37 °C, 10 min, 1 h), and plasma was separated from whole blood samples through centrifugation (2,000 xg, 10 min). Concentrations of AT and BE in both plasma produced from spiked whole blood and spiked plasma) were measured using HPLC as described above. The assay was done three times.
Blood-to-plasma ratios (B:P) of AT and BE were expressed as RBC-to-plasma partitioning ratio (KRBC/PL), which is the ratio of the concentration of the compound in RBC (CRBC) over that in the equilibrating plasma (CPL):
Where, H = Hematocrit
IRef PL: Peak area of the analyte in the reference plasma.
IPL: Peak area of the analyte in plasma separated from whole blood.
Results and Discussion
Plasma protein binding
The distribution of the drug into target tissues depends on the degree of plasma protein and tissue binding. The unbound fraction (fu,p) represents the fraction of a compound that is not bound to blood components (e.g., plasma proteins and blood cells) and is available to interact with target molecules in the target tissues, such as receptors, ion channels, enzymes, macromolecules, and nucleic acids, as well as metabolizing enzymes and other molecules involved in the ADME processes. Therefore, it determines drug concentration at the site of action, and thus clinical efficacy. As the fraction of unbound drugs influences renal glomerular filtration and hepatic metabolism, a change in unbound drug concentration would affect the volume of distribution and total clearance of the drug. Both pharmacokinetic parameters are the key parameters for determining drug dosage regimens. In addition, when the degree of plasma protein binding (%PPB) is high, small protein binding changes may significantly impact fu,p, and consequently, the therapeutic efficacy [15]. Hence, unbound concentrations should be used to estimate drug pharmacokinetic parameters and PK-PD analysis for dose optimization.
The ultracentrifugation method is commonly employed to determine the unbound fraction of compounds in plasma, especially lipophilic compounds. The results demonstrated that the mean fu of AT and BE at all three concentrations (2.5, 5, and 10 ug/ml) were 0.04 and 0.05, respectively (Table 1). Plasma protein binding of both compounds was shown to be relatively high (PPB >90%), suggesting their high binding affinity to plasma proteins. Compound lipophilicity (LogP or LogD) may explain drug molecules' protein binding, Vd, and other distribution properties. The current in vitro study generally supports our results by in silico prediction (unpublished data), which showed fu values of 8.51% and 5.92% for AT and BE, respectively. The distribution of both compounds is, thus, likely to be limited to the systemic circulation (plasma and blood cells) rather than being extensively distributed into other tissue compartments in the body [16, 17]. Previous studies in animals and humans showed low and short systemic bioavailability of AT and BE after oral administration [7-12].
AL and BE are basidic compounds [13] and tend to bind with alpha 1-acid glycoprotein [18]. This plasma protein is an acute phase protein of which the concentration can be affected by various pathophysiological conditions or diseases, including stress, surgery, bacterial/viral/parasitic infections, liver or kidney dysfunction, and cancer [19, 20]. Consequently, this change may result in the alteration of the unbound fraction of the compound.
Table 1: Unbound fractions fu and plasma protein binding (%) of atractylodin (AT) and β-eudesmol (BE) in human plasma (n=10). Data are presented as mean+SD of three independent experiments
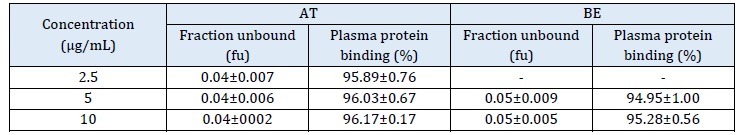
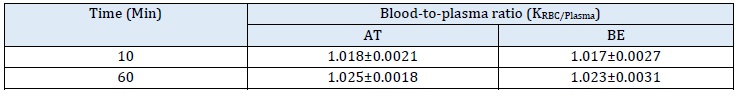
Table 2: Blood-to-plasma ratio (Kb/p) of atractylodin (AT) and β-eudesmol (BE) in human plasma (n = 10). Data are presented as mean+SD of three independent experiments
Furthermore, a relatively high protein binding nature of AT and BE may increase the risk of drug-drug interactions through plasma protein binding displacement. For highly bound compounds (>95%), clinical ex vivo protein-binding studies should be performed, and whole blood rather than plasma should be the matrix of choice for measurement of free drug concentrations.
Blood-to-plasma concentration ratios
Several drugs have the potential to bind or penetrate RBCs. RBCs comprise at least 90% of cellular space in blood, so this binding could significantly impact drug clearance, leading to inaccuracies in estimating pharmacokinetic parameters. The partitioning of drugs into RBCs primarily occurs through passive diffusion across the cell membrane, but may also involve channels and carriers, making the process concentration-dependent and saturable [21]. Rather than whole blood, plasma is a commonly used biological matrix for pharmacokinetic studies of drugs or candidate compounds. It is critical to realize the difference between their plasma and whole blood concentrations and the clinical consequences. Failing to consider the potential for a drug to sequester into RBCs may lead to a misinterpretation of pharmacokinetic data and provide an overestimation of drug clearance. The blood-to-plasma (B:P) ratio values obtained from the present study will be further used to adjust concentration-time profiles and clearance, ensuring the accuracy of the pharmacokinetic parameter estimation [22, 23].
The present study evaluated blood-to-plasma partitioning of AT and BE based on blood-to-plasma partition coefficients (KRBC/Plasma). AT and BE Concentrations were determined by HPLC in both plasmas produced from the whole spiked blood and the spiked plasma.
The mean KRBC/Plasma of AT and BE were slightly increased from 1.01 to 1.025 and 1.017 to 1.023, when the incubation time was increased from 10 to 60 min (Table 2). The results indicate that AT and BE distribution into RBC undergoes saturation at a slightly higher accumulation in RBCs compared to plasma (KRBC/Plasma >1.0) [24]. This is explained by the lipophilic nature of AT and BE (LogD >3.5) [9]. Both plasma and whole blood can be used as the matrix of choices for the estimation of pharmacokinetic parameters of AT and BE. The previous study used AT and BE concentrations in plasma to determine the PK parameters [12]. It is recommended that [23], if the B:P ratio is approximately 1.0, either whole blood or plasma is the matrix of choice for pharmacokinetic evaluation. On the other hand, if the ratio is less than 1.0 and greater than 1.0, plasma and whole blood are, respectively, the matrix of choice. The antimalarial drug chloroquine accumulates in blood cells (RBCs, WBCs, and platelets), leading to high B:P ratios. Chloroquine concentrations in whole blood are approximately 3 to 10 times higher than in plasma. Due to the elution of the drug into the serum during clotting, serum chloroquine concentrations are higher than those in plasma. The large volume of distribution of chloroquine indicates extensive tissue binding, but it is weakly bound in plasma (46-74% in healthy subjects) [26]. RBCs concentration of digoxin is 2-6 times higher than in plasma [27], and carbamazepine is 11-23% of plasma concentrations [28]. Thrombocytes (platelets) have been shown to bind another strongly basidic drug, imipramine [29]. For chlorthalidone, whole blood concentration is ten times higher than in plasma [30].
Conclusion
The high partitioning in blood circulation and high plasma protein binding of AT and BE in RBCs may limit the extent of their distribution and delivery to the target tissues (bile ducts). Either plasma or whole blood can be used as the matrix of choice for pharmacokinetic studies. The information provides essential input parameters in the pharmacokinetic models to predict optimal dose regimens for AL in clinical studies.
Conflict of Interest
Both authors declare no competing financial interests or personal relationships.
Funding
The study was supported by the National Research Council of Thailand (Research Team Promotion grant No. 820/2563) and the Thailand Science Research and Innovation Fundamental Fund Fiscal year 2024.
Authors’ Contributions
Both authors contributed to data analysis, drafting, and revising of the paper and agreed to be responsible for all aspects of this work.
ORCID
Kesara Na-Bangchang
https://orcid.org/0000-0001-6389-0897
HOW TO CITE THIS ARTICLE
Anurak Cheoymang, Kesara Na-Bangchang. Plasma Protein Binding and Blood-to-Plasma Ratios of Atractylodin and -Eudesmol, the Bioactive Compounds from Atractylodes lancea (Thunb.) D.C. J. Med. Chem. Sci., 2024, 7(1) 132-139.
)