Document Type : Original Article
Authors
1 Department Pharmaceutical Chemistry, College of Pharmacy, University of Duhok, Duhok, Kurdistan
2 Department of Pharmaceutical Chemistry, College of Pharmacy, Hawler Medical University, Erbil, Kurdistan
3 Department of Chemistry, College of Science, University of Mosul, Mosul, Iraq
4 Department of Pharmaceutical Chemistry, College of Pharmacy, University of Mosul, Mosul, Iraq
Abstract
Nonsteroidal anti-inflammatories (NSAIDs), are very effective agents in relieving mild to moderate pain and inflammation by inhibiting two isoforms of prostaglandin G-H synthetase (I and II). In the present work, anthranilic acid derivatives' electronic and physicochemical properties are reported utilizing quantum chemical calculations that use the density functional theory (DFT), which forecast physicochemical properties. To clarify the type of chemical composition, drug-likeness, and cyclooxygenase inhibitor, ADME and molecular docking were used. The molecule was highly electrophilic and relatively stable from a quantum chemical computation perspective. The contour maps of HOMO-LUMO and molecule electrostatic potential were examined to display the charge density distributions that might be related to the biological activity.
Graphical Abstract
)
Keywords
Main Subjects
Introduction
Non-steroid and anti-inflammatory drugs are non-specific inhibitors of certain manifestations of inflammation [1]. Most of the FDA-approved non-steroidal anti-inflammatory drugs (NSAIDs), whether they are Salicylates [2], Anthranilic acid [3], Arylindoic acid [4], and Oxicame [5] (Scheme 1), possess common structural features of an acidic or amine of an aromatic or heterocyclic ring, and an additional center of lipophilicity mostly aromatic. Their mechanisms are non-selective, inhibiting both COX-I and COX-II isoforms [6, 7].
Density-functional theory (DFT) is a computational quantum mechanical method widely used in modern medicinal chemistry to suggest the electronic 3D atoms or molecule structures compute the ground state energy in realistic models of bulky molecules and their surfaces [8, 9]. On the other hand, a valuable and popular tool in drug design is molecular docking. Predicting the binding affinity between a small molecule (ligand) and a macromolecule (receptor), which is crucial for medication development, is a computer process. Recently, a molecular docking research on the assessment of prospective antibacterial medicines was released. Some scoring formulae forecast the ligand's biological and complimentary activities. Mostly docking scores are more significant than being in a precise place [10].
The majority of NSAID moieties are chemically made up of carboxylic functional groups, which may be one of the causes of mucosal membranes' direct injury. The ester, amides, amine, and anhydride derivatives completely mask the carboxylic groups and decrease the main direct effect of gastrointestinal (GI) ulceration and irritation [11]. Recently, Dana Ameen et al. has been paying special attention to the diclofenac derivatives which exhibited no toxicity, and the preservation of stomach wall mucus may be the cause of gastroprotective effect [12].
This work aims to predict the theoretical density functional theory, docking, and pharmacokinetic parameters of the selected new anthranilic compounds.

Materials and Methods
Density functional theory (DFT): The chemical structures of anthranilic acid derivatives and their models have been drawn using two-dimensional ChemDraw ultra version 11.0. Each molecular structure was transferred using the Chem 3D-ChemBioOffice software version 16.0.0.82 and a systematic energy minimization (level: ultra). The optimization process began with molecular mechanics calculations (MM2), and then MMFF94 methods were utilized to obtain a value of the root mean square (RMS) gradient that was less than 0.1 kcal/mol to arrive at a negative sign for the heat of formation and a positive sign for frequency, the minimization procedure was continued using semi-empirical calculations, including the Austin Model 1 (AM1) and Parameterized Model 3 (PM3) approaches. The energy minimizations were carried out for density functional theory (DFT) calculations using DFT at the B3LYP level with a 6-311G basis set until the minimal RMS gradient of 0.1 was attained [13]. Depending on the type of used descriptor, the estimation of descriptors was performed by the Gaussian 03w software utilizing DFT, Hartree-Fock ab initio (HF), and PM3 techniques (closed-shell MOs) [14].
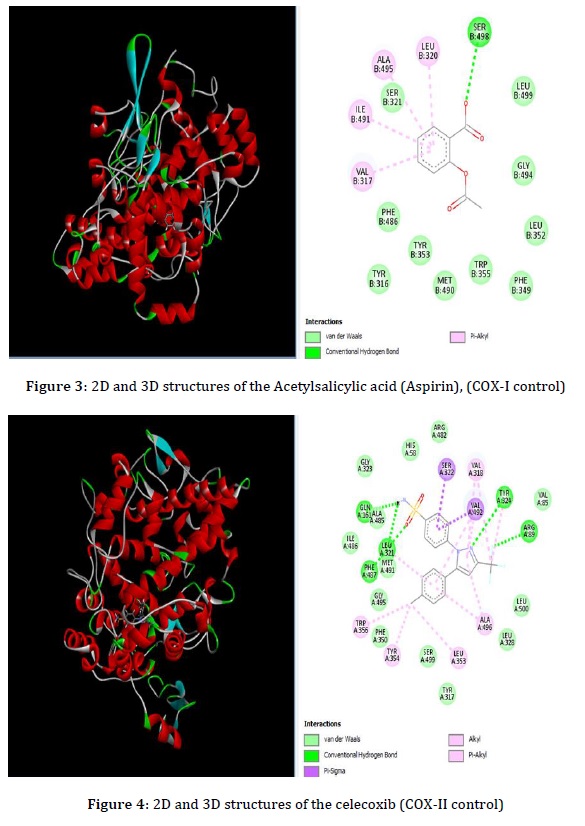
Docking Study: To determine the binding modes of the most active drugs, an in silico technique based on structure was used to assign 2, 6, 9, and 10 to prostaglandin G-H synthetase I enzyme active sites, which were crystallized with cyclooxygenase-1 reference drug (Acetylsalicylic Acid), while the binding modes of active compounds 5 and 9 to the prostaglandin G_H synthetase II enzyme active site, crystallized with the reference drug (celecoxib). Both were retrieved from the Protein Data Bank server (http://www.pdb.org). The 2D and 3D structures of the compounds and binding scores are fixed in Table 1 [15].
Prediction of Pharmacokinetic parameters: The physicochemical parameters ADME [absorption, distribution, metabolism, and excretion] of twenty anthranilic acid derivatives were calculated using sever www.swissedme.com as shown in (Table 2) [16, 17].
Results and Discussion
The reliability of 20 compounds was calculated depending on the development of approximations for exchange-correlation energy function, the 3D structures of HOMO and LUMO are displayed in Figure 1.
Quantum chemical calculations
Oopmans' theorem represents a theoretical method for connecting chemical activities of molecular structures to their electrical characteristics [18]. A molecule's reactivity could be determined using quantum chemical descriptors derived from Koopmans' theorem, such as ionization potential (I), electronic affinity (A), chemical potential (µ), hardness (η), softness (σ), electronegativity (χ), and electrophilicity (ω). These parameters’ mathematical definitions are listed in Table 2. The examined compound's HOMO and LUMO energies, any related chemical characteristics, are listed in Table 2. The HOMO and LUMO energy hole serve as a gauge for the kinetic stability of the molecule. High reactivity is linked to a short HOMO-LUMO hole, whereas high chemical stability is attributed to a high energy gap value [19, 20]. Electronic affinity (A) is the energy released when a molecule in its ground state captures an electron, and ionization potential (I) is the energy needed to remove an electron from a molecule's ground state. While a molecule with a high electronic affinity value is more likely to take electrons, one with a high ionization energy value suggests chemical stability. The examined compound exhibits good chemical stability, according to Table 2, because of its wide energy gap, high ionization potential, and weak electron affinity [21].


Docking study
For molecular docking computation, Mcule Docking online was used to predict the specific interactions between the ligand (compounds 1-20) and target proteins, including their binding affinities (Prostaglandine G_H synthetase). The ligand-protein complex was created and predicted the protein's active location where ligand is in its optimal shape. The Discovery Studio Visualizer software 2021 was used to extract and display the ligand-protein interactions Table 3 [22].
Prediction of ADME
Information about pharmacokinetics is now frequently found on numerous well-known websites. In this experiment, the ADME and drug-likeness qualities were assessed using the Swiss ADME. To give safety considerations for a novel drug on which risk-based evaluations may be made, it is helpful to examine and explain how pharmacokinetic processes take place through the characterization of absorption, distribution, metabolism, and excretion (ADME) features (Table 4). 20 compounds are small molecules and can apply the Lipinski rule (Ro5). This rule importance in identifying clinically meaningful pharmacokinetic drug-likeness still seems to be valuable today. Calculations of the selective compounds 1-20 (COX-I and COX-II) [23, 24] are indicated in (Table 3).
Nowadays, it is standard practice to use computational results in experimental research on orbitals and surfaces in 3 dimensions because modern DFT simulation algorithms for solid-state calculations can calculate a wide range of structural, chemical, spectroscopic, and thermodynamic properties. The title molecule was optimized using the Gaussian 03 software suite, which represents the DFT approach in the model of B3LYP level with a 6-311** basis set [25]. Utilizing the Gauss View 6 graphical user interface, the output data, including the optimized geometry, HOMO-LUMO, and MEP were made visible. It is demonstrated that Koopmans' theorem for a large molecule system can be expressed as follows in density functional theory (DFT): The highest occupied molecular orbital (HOMO) energy minus the Coulomb electrostatic energy of removing an electron from the system equals the ionization energy of the system as well as other parameters, as shown in Equations (i-viii) [25, 26].

The simple calculation was done to calculate all other physical parameters such as ionization potential (I, electronic affinity (A), hardness (η), softness (σ), chemical potential (µ), electronegativity (χ), and electrophilicity (ω). These parameters’ mathematical definitions are listed in (Table 2).
The use of theoretical docking programs to predict the interaction between the receptor (enzyme) and the ligand (molecule) to form a more stable receptor-ligand complex inhibitor. In the present work, the interactions of prostaglandin G-H synthetase I (PDB:) protein with twenty anthranilic derivative compounds were investigated. The 2D and 3D representations of the compound and two controls, acetylsalicylic acid (Aspirin) as (COX-I control), and celecoxib as (COX-II control), as shown in Figures 2, 3 and 4.
There are numerous interaction centers, including the sulfonamide amino acid Leu 321 NH and oxygen atoms of the sulfonyl group, as well as other hydrogen bonds between Tyr 324 and Arg 89 to the pyrazole ring. Other weak bonds are pi- sigma between the phenyl ring, Ser 322, and Val 492 [14].
The tested twenty compounds showed binding energy ranging from -7.8 to -9.8 Kcal/mole (Table 1). The compounds 2, 6, and 9 showed a solid crucial score compared with the acetylsalicylic acid (control). This indicates they fit well in the binding pocket on the prostaglandin G_H synthetase in forming a stable inhibitor protein complex. The compounds 11-20 showed a weak interaction with the enzyme prostaglandin G_H synthetase II.
In addition, all anthranilic compounds (ligands) had their physicochemical characteristics evaluated and predicted by RO5 [19] using in silico computational techniques, raising the possibility that they could be orally bioavailable medicines. All selected compounds obeyed the Lipinski rule except compounds 1, 11, 12, and 16. Furthermore, all compounds except compounds 10, 14, 17, and 20 are non-blood brain barrier (BBB), as presented in Table 1.

Conclusion
In this study, we represented the DFT, docking using prostaglandin G-H synthetase I enzyme, pharmacokinetic properties (ADME), and drug likeness prediction of twenty selected anthranilic derivatives. New series of anthranilic acid derivatives were designated as cyclo oxygenase inhibitors, docking, and theoretical study. Their physicochemical properties were thoroughly discussed. More research is required to manufacture active ingredient and assess its anti-inflammatory efficacy.
Acknowledgements
The authors would like to thanks university of Duhok, Hawler Medical University, University of Mosul for provided facilities to complete this article.
Disclosure Statement
No potential conflict of interest was reported by the authors.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Authors' contributions
All authors contributed to data analysis, drafting, and revising of the paper and agreed to be responsible for all the aspects of this work.
ORCID
Karam S. Atrushi
https://orcid.org/0000-0001-7754-0329
Dana M. Ameen
https://orcid.org/0000-0001-8391-2053
Shaymaa H. Abdulrahman
https://orcid.org/0000-0001-8270-0892
Faris T. Abachi
https://orcid.org/0000-0003-3389-877X
HOW TO CITE THIS ARTICLE
Karam S. Atrushi, Dana M. Ameen, Shaymaa H. Abdulrahman, Faris T. Abachi. Density Functional Theory, ADME, and Molecular Docking of Some Anthranilic Acid Derivatives as Cyclooxygenase Inhibitors. J. Med. Chem. Sci., 2023, 6(9) 1943-1952
)