Document Type : Original Article
Authors
- M. Nizam Zulfi Zakaria 1
- Ahmad Fariduddin Aththar 1
- Michelle Fai 1
- Syeftyan Muhammad Ali Hamami 1
- Viol Dhea Kharisma 2, 3
- Ahmad Affan Ali Murtadlo 2, 3, 4
- Arif Nur Muhammad Ansori 2, 3, 4, 5, 6, 7
- Teguh Hari Sucipto 8
- Rahadian Zainul 9
1 Department of Biology, Faculty of Mathematics and Life Sciences, Universitas Brawijaya, Malang, Indonesia
2 Generasi Biologi Indonesia Foundation, Gresik, Indonesia
3 Department of Biology, Faculty of Science and Technology, Universitas Airlangga, Indonesia
4 Uttaranchal Institute of Pharmaceutical Sciences, Uttaranchal University, Dehradun, India
5 Division of Research and Development, CV Jalan Tengah, Pasuruan, Indonesia
6 European Virus Bioinformatics Center, Jena, Germany
7 Postgraduate School, Universitas Airlangga, Surabaya, Indonesia
8 Dengue Study Group, Institute of Tropical Disease, Universitas Airlangga, Surabaya, Indonesia
9 Department of Chemistry, Faculty of Mathematics and Life Sciences, Universitas Negeri Padang, Padang, Indonesia
Abstract
The Chikungunya virus (CHIKV), a zoonotic virus transmitted through mosquito bites, can cause dengue-like fever in humans. Despite the lack of specific clinical treatments and vaccines, there has been limited attention given to CHIKV in recent decades. This study utilized an immunoinformatics approach to design a potential multi-epitope vaccine for CHIKV. The CHIKV multi-epitope vaccine (CHIKV-MEV) was created by combining predicted linear B-cell lymphocyte (LBL), cytotoxic T-lymphocyte (CTL), and helper T-lymphocyte (HTL) epitopes targeting the viral envelope glycoprotein E1/E2 of CHIKV. The selection of each epitope was based on parameters such as antigenicity, immunogenicity, toxicity, and allergenicity. These selected epitopes were incorporated to generate refined and validated 3D models of CHIKV-MEV. Molecular docking simulations were performed to assess the interaction between the generated 3D model of CHIKV-MEV and TLR-1/2. Immune response simulations and population coverage analysis were conducted to evaluate the potential effectiveness of the vaccine. The proposed CHIKV-MEV consists of 439 amino acids, encompassing 18 epitopes, and exhibits predicted properties of being antigenic, immunogenic, non-allergenic, and non-toxic. The binding energy of -1079.0 kcal/mol indicated that CHIKV-MEV can interact with TLR-1/2, leading to immune responses. Immune response simulations of CHIKV-MEV demonstrated an increase in immunoglobulin levels, as well as population of LBL, CTL, and HTL, and cytokine levels associated with the defence against viral infections. Furthermore, based on compatibility with human leukocyte antigen (HLA), CHIKV-MEV potentially covers 96.25% of the global population. This research contributes to the development of a globally applicable multi-epitope peptide-based vaccine against CHIKV, supported by comprehensive in vitro and in vivo studies.
Graphical Abstract
)
Keywords
Introduction
Chikungunya is an infectious disease caused by the chikungunya virus (CHIKV), which is transmitted to humans through the bite of Aedes mosquitoes carrying CHIKV RNA [1]. Aedes aegypti (the yellow fever mosquito) and Aedes albopictus (the Asian tiger mosquito) are potential mosquito vectors of chikungunya, known to carry the RNA virus [2]. CHIKV belongs to the Alphavirus genus of the Togaviridae family and serves as the causative agent of the disease. Acute symptoms of chikungunya mainly include fever and joint pain, leading to arthralgia and significantly impacting individuals, although the disease itself is not fatal [3]. Chikungunya has a relatively high prevalence rate in Indonesia [4].
The discovery of the chikungunya virus (CHIKV) dates back to 1952 in Tanzania, where it was identified as the causative agent of a pathogenic epidemic in humans. The virus was isolated from the serum of infected patients during that time [5]. The chikungunya virus is widespread in tropical areas of sub-Saharan Africa, such as the continents of Asia, Africa, and North America [6]. Based on the World Health Organization (WHO) data (2022), it shows that the spread of chikungunya since 2004 until now has spread rapidly in more than 110 countries on the continents of Africa, Asia, Europe, and the Americas. Data as of July 26, 2023 shows that there have been 300,000 new cases and more than 300 deaths reported from all over the world, dominated by the Americas from Brazil (192,822), Paraguay (101,963), Argentina (1.593), Bolivia (1.311), and in Asia (Thailand) (598 cases) (European Centre for Disease Prevention and Control, 2023). As of 2019, there have been as many as 1.9 million cases of chikungunya in Asia, of which 5,042 cases were found spread across 20 provinces in Indonesia, and data on extraordinary events of chikungunya patients starting from 2013 to 2018 were recorded in as many as 149,526 cases [7, 8].
Thus, along with the increase in the number of people with chikungunya disease in the current decade, vigilance and preventive efforts are needed to reduce the burden in the treatment management.
However, reality shows that, until now there has been no treatment therapy or anti-CHIKV vaccine that has an official license. So far, the treatment carried out is only through palliative care, namely the use of analgesics, antipyretics, and NSAIDs, which aim to relieve chikungunya symptoms such as fever and joint pain, and prevention is carried out only through controlling chikungunya vectors [9]. A number of strategies to overcome chikungunya have been carried out through several studies, one of which is Clayton's (2016) [10] research, which shows that a number of CHIKV antivirals can stimulate the humoral immune response pathway by controlling the infection process, where monoclonal antibodies will play a role in protecting the body by slowing down the infection process. This indicates that the most effective treatment strategy for chikungunya infection can be achieved through immunotherapy through anti-CHIKV vaccines. Vaccines were also chosen because vaccines can protect against all types of CHIKV and will also provide lifelong immunity, and treatment with vaccines can be carried out quickly in certain populations, so there is a great opportunity to minimize the risk of increasing the occurrence of chikungunya epidemics [11].
To expedite and streamline the development of anti-CHIKV vaccines, modern vaccine design methods employing immunoinformatics and bioinformatics technology approaches are required, as traditional approaches are time-consuming and costly. Protein vaccines, which utilize small portions of pathogenic proteins as amino acid sequences, necessitate the identification of potential epitopes or antigenic determinants that serve as specific targets for antibody recognition, thereby eliciting B-cell and T-cell immune responses [12].
Referred to as a multi-epitope vaccine, this vaccine design approach involves the use of filters based on experimental data to select the most suitable antigenic epitopes for further evaluation in experimental settings, as described by Battacharya et al. (2022) [13].
The multi-epitope vaccine design method serves as a predictive tool for researchers and scientists to guide experimental research on the chikungunya virus pathogen.
The structural proteins of CHIKV encompass a capsid protein, two envelope glycoproteins (E1 and E2), and two small cleavage products (E3 and 6K), as reported by Petitdemange et al. (2015) [14]. According to Hasan et al. (2018) [15], the envelope glycoproteins E1 and E2 play crucial roles in facilitating viral infection by mediating viral entry into target cells through host endocytosis processes. Concerning the functional significance of Envelope 1 (E1) and Envelope 2 (E2), both proteins hold potential as target materials for the design of multi-epitope vaccines.
Therefore, it is imperative to conduct a study focused on the design of multi-epitope vaccines targeting the Chikungunya virus. In this particular study, the design of the CHIKV multi-epitope vaccine (CHIKV-MEV) involved the integration of predicted epitopes from linear B-lymphocytes (LBL), cytotoxic T-lymphocytes (CTL), and helper T-lymphocytes (HTL), all based on the effective epitope vaccine antigen provided by the CHIKV E1/E2 viral envelope glycoprotein and utilized in CHIKV vaccines.
Materials and Methods
This study was conducted in the Laboratory of Computational Biology and Bioinformatics, Biology Department, Faculty of Mathematics and Life Science, Brawijaya University, Malang City, East Java, Indonesia. The methods for designing CHIKV-MEV were done by several steps, including CHIKV envelope glycoprotein retrieval, lymphocytes epitopes prediction, vaccine assembly, 2D and 3D model construction, immune response simulation, and population coverage analysis.
Data mining of CHIKV E1/E2 envelope glycoprotein
The amino acid sequences of the E1 and E2 glycoproteins were obtained in Fasta format from the GenBank web server (https://www.ncbi.nlm.nih.gov/genbank/), while the crystallized structure of the E1 and E2 glycoproteins was obtained in pdb format from the Protein Data Bank (PDB ID: 2XFB) (https://www.rcsb.org/). The length of E1 and E2 glycoproteins were 391 and 334 amino acids, respectively. The three-dimensional structure of those glycoproteins was validated by electron microscopy with 9 Å resolution (Voss et al., 2010) [16].
Data mining of potential linear B-lymphocyte epitopes
The Immune Epitope Database and Analysis Resource web server (http://tools.iedb.org/bcell/) was utilized to predict the linear B-lymphocyte (LBL) epitopes. The Bepipred Linear Epitope Prediction 2.0 method, which employs a Random Forest algorithm trained on epitopes and non-epitope amino acids derived from crystal structures, was employed for this purpose.
Accordingly, a sequential prediction smoothing process was implemented. Residues that exceed a specified threshold (>0.5) were anticipated to be constituents of an epitope [17].
T- and B-lymphocyte epitopes screening
All predicted epitopes underwent screening to assess various properties. The antigenicity property was determined by employing the VaxiJen 2.0 web server (http://www.ddg-pharmfac.net/vaxijen/) with a threshold of zero (0.0). The antigenicity score was calculated based on the physicochemical characteristics of proteins, as described by Doytchinova et al. (2007) [18].
To predict T- and B-lymphocyte epitope allergenicity, the AllerTOP web server (http://www.ddg-pharmfac.net/AllerTOP/) developed by Dimitrov et al. (2014) [19] was utilized. In addition, the toxicity of each epitope was predicted using the ToxinPred2 web server (https://webs.iiitd.edu.in/raghava/toxinpred/) according to Gupta et al. (2013) [20]. Furthermore, the immunogenicity property of cytotoxic T-lymphocyte epitopes was assessed using the MHC I immunogenicity prediction tool (http://tools.iedb.org/immunogenicity/) (Calis et al. 2013) [21].
Vaccine construction, validation, and refinement
The chosen epitopes were combined and assembled along with the adjuvant and linkers sequence. The adjuvant sequence of 50S ribosomal protein L7/L12 (UniProt ID: P9WHE3) was obtained from the UniProt database (https://www.uniprot.org/). The attachment of LBL epitopes to the adjuvant was accomplished using the EAAAK linker, followed by attachment to cytotoxic and helper T-lymphocyte epitopes using KK, AAY, and GPGPG linkers [22]. The resulting multi-epitope vaccine sequence was then used to generate secondary and tertiary structures using PDBsum (http://www.ebi.ac.uk/thornton-srv/databases/pdbsum/) and I-TASSER (https://zhanggroup.org/I-TASSER/), respectively [23, 24]. Subsequently, the initial 3D structure was refined using the GalaxyRefine tool (https://galaxy.seoklab.org/) to enhance the quality of the peptide structure [25]. The MolProbity web server (http://molprobity.biochem.duke.edu/) was employed to validate the quality of the resulting vaccine's 3D structure [26].
Molecular docking simulation
To assess the efficacy of the vaccine, a molecular docking simulation was performed by applying it to the TLR-1/2 receptor (PDB ID: 2Z7X) using the Cluspro2.0 web server (https://cluspro.bu.edu/) as described by Comeau et al. (2004) [27]. Subsequently, the molecular interaction between the TLR-1/2 receptor and the vaccine was analysed using the PDBsum web server (http://www.ebi.ac.uk/thornton-srv/databases/pdbsum/) to evaluate parameters such as binding affinity, amino acid residues involved, and the type of interactions, following the methodology outlined by Laskowski (2001) [28].
Immune response simulation
The prediction of the vaccine's ability to stimulate an immune response was performed using the C-ImmSim web server (https://kraken.iac.rm.cnr.it/C-IMMSIM/). This web server employs a position-specific scoring matrix (PSSM) approach to identify immunological epitopes and assess immune interactions, as outlined by Rapin et al. (2011) [29].
Population coverage analysis
The analysis of population coverage for the specific vaccine was conducted using the dedicated tool provided on the IEDB web server (http://tools.iedb.org/population/). The prediction of coverage percentage worldwide was determined based on MHC-I and MHC-II epitope data, as described by Bui et al. (2006) [30].
Results and Discussion
Virulence capability of CHIKV E1/E2 envelope glycoprotein
Chikungunya viruses (CHIKV) have the ability to virulate by binding to receptor-binding glycoproteins in infected hosts. CHIKV glycoproteins are the main determinants of CHIKV virulence. The CHIKV virulence cycle begins with the fusion of viral particles into the cell membrane through viral attachment. The process is carried out by endocytosis, facilitated by Envelope 1 (E1) and Envelope 2 (E2), to enter the target cell. The E1 protein is involved in mediating the fusion process between the viral membrane and the host cell, whereas the E2 protein acts as a viral spike that binds to the specific target receptor (Hassan et al. 2018) [31]. Extensive in vitro and in vivo studies, employing mosquito and mouse models, have demonstrated the critical role of both E1 and E2 glycoprotein domains in determining the virulence capacity of alphaviruses, including CHIKV. These studies have revealed the interplay between E1 and E2 domains, which is essential for the assembly and replication of the CHIKV virus with host target proteins [32]. Notably, the E1 glycoprotein participates in a blocking pathway by immobilizing CHIKV on the host plasma membrane surface, thereby inducing an inflammatory response that effectively inactivates the virus. As for CHIKV E2, when the surface of the two domains (domains A and B) is exposed to E2 with GAGs, it will bind to cells to inhibit CHIKV infection by 90% [33]. In addition to functioning for the attachment of CHIKV entry, glycoproteins also have a role as an immunogenicity response to CHIKV-infected cells [34]. Thus, receptor-binding E1 and E2 proteins regulate the activation of fusion proteins prematurely by structurally covering them so that the dissociation of receptor-binding proteins from membrane fusion proteins can be blocked as an inhibition of virus entry [35]. Based on this description, the two glycoproteins, Envelope 1 (E1) and Envelope 2 (E2) can be used as effective specific target materials for anti-CHIKV multi-epitope vaccine design.
Prediction of linear B-lymphocyte (LBL) epitopes
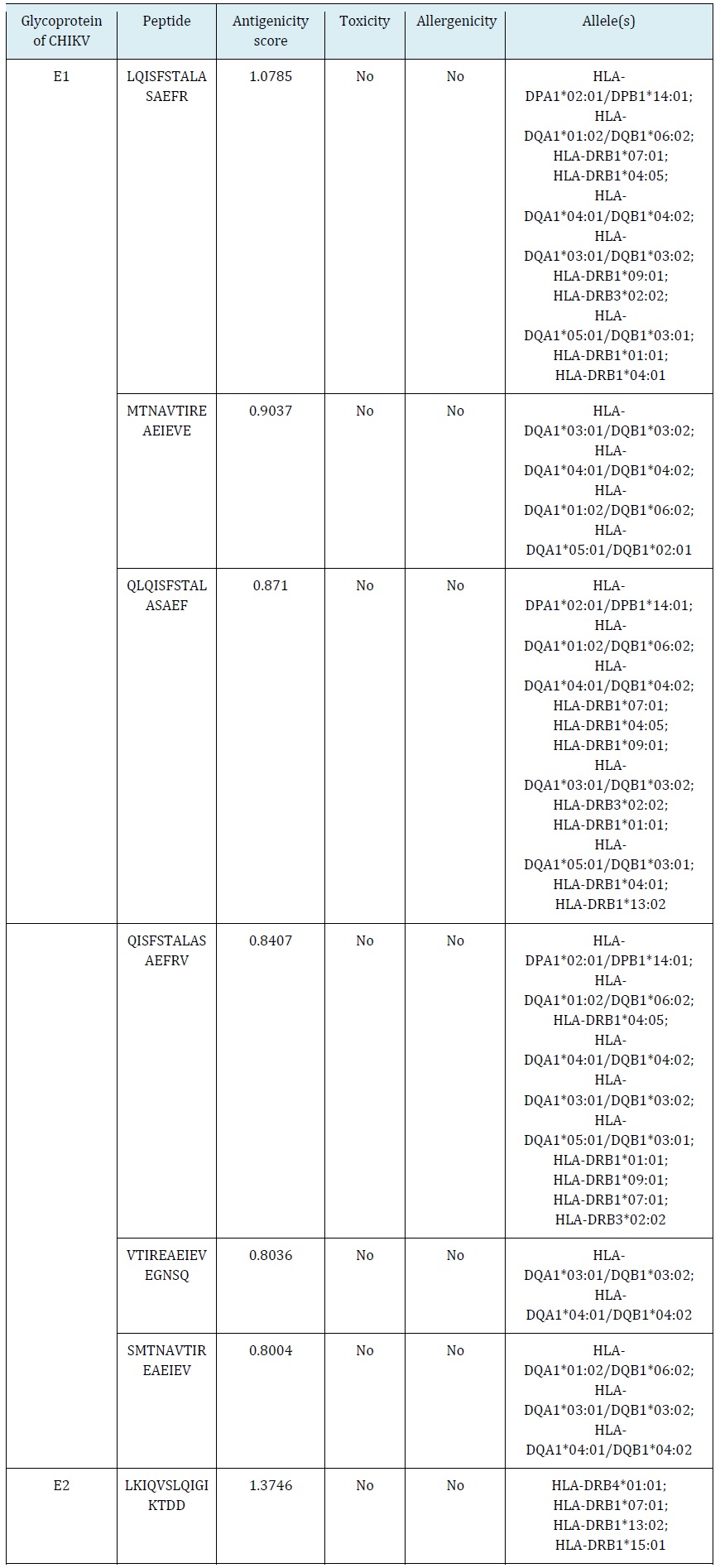
Two potent linear epitopes were identified from each CHIKV E1 and E2 by the IEDB database. They were all confirmed as antigenic, non-toxic, and non-allergenic epitopes by using VaxiJen v2.0, Toxin Pred, and AllerTOP v2.0, respectively (Table 1).
Prediction of cytotoxic T-lymphocyte (CTL) epitopes
Five potent epitopes from each CHIKV E1 and E2 recognized by CTL due to interaction with predicted MHC class-1 alleles. Their antigenicity scores were > 0.8 based on VaxiJen v2.0. Furthermore, they were all considered immunogenic, non-toxic, and non-allergenic epitopes using IEDB analysis resources, Toxin Pred, and AllerTOP v2.0, respectively (Table 2).
Prediction of Helper T-Lymphocyte (HTL) Epitopes
CHIKV E1 and E2 had seven epitopes recognized by HTL through their interaction with MHC class-2 alleles. The candidates incorporated six E1 epitopes and a single E2 epitope with antigenicity scores > 0.8 based on VaxiJen v2.0. Hence, they were considered non-toxic and non-allergenic using Toxin Pred and AllerTOP v2.0, respectively (Table 3).
Table 1: Predicted linear epitopes for B-lymphocyte recognition

Table 2: Predicted epitopes for cytotoxic T-lymphocyte (CTL) recognition

Table 3: Predicted epitopes for Helper T-Lymphocyte (HTL) recognition
Multi-epitopes vaccine assembly
Our proposed CHIKV vaccine comprises eighteen epitopes, composing four linear B-cell lymphocyte (LBL) epitopes, seven cytotoxic T lymphocyte (CTL) epitopes, and seven helper T lymphocyte (HTL) epitopes. Three CTL epitopes (GQFGDIQSR, TALASAEFR, and QVSLQIGIK) merged due to matching sequences found in one LBL epitope and two HTL epitopes, which have underlined. Each epitope has been designated with an asterisk, indicating its source region of the CHIKV glycoprotein. The vaccine sequence also contains an adjuvant (50S ribosomal protein L7/L12), various linkers, and a histidine tag (HHHHHH). EAAAK, KK, AAY, and GPGPG linkers connect the primary LBL epitope to the adjuvant, LBL epitopes, CTL epitopes, and HTL epitopes, respectively. Histidine tag appendage facilitates protein synthesis and enhances expression in E. coli vector (Figure 1)
Vaccine’s secondary structure visualization
The secondary structure visualization of the vaccine was conducted using the PDBsum server, as depicted in Figure 2. The protein's actual properties were analysed, and based on this information, the secondary structure was extrapolated. The extrapolation results revealed that the protein comprises 3 sheets, 12 strands, 11 helices, 69 beta turns, and 10 gamma turns.
Vaccine’s tertiary structure visualization
To obtain specific modelling of the chimeric tertiary structures, we employed the I-TASSER server.

Figure 1: (A) Schematic construction of proposed multi-epitope CHIKV vaccine and (B) final sequence of multi-epitope CHIKV vaccine including adjuvant, linkers, and histidine tag

Figure 2: CHIKV-MEV secondary structure visualization
The modelling process involved utilizing the top five threading templates, which were chosen based on their highest coverage values. Subsequently, models derived from the threading templates with the highest coverage values were selected for subsequent refinement in this study.
Tertiary structure validation and refinement
To obtain the most accurate tertiary structure model of the CHIKV vaccine, the 3D model generated by I-TASSER underwent refinement using the Galaxy Refine tool, yielding five distinct vaccine models. Among these models, the first model exhibited the most promising characteristics, including a GDT-HA score of 0.9436, an RMSD value of 0.43, and a MolProbity score of 2.283. The Ramachandran score was expected to be 89.5%, while the clash score was determined as 16.5, and the poor rotamer score was 0.6. To assess the quality of the refined model, evaluation was conducted using the MolProbity server, resulting in the generation of a Ramachandran plot (Figure 3). Prior to refinement, 67% (293/437) of all residues resided within the favoured region, and 89.9% (393/437) of residues fell within the allowed region, with 44 outliers identified. Following refinement, a significant reduction in outliers was observed, leaving 14 outliers in the final model.
Moreover, there was a notable improvement, with 89.5% (391/437) of all residues occupying favoured regions and 96.8% (423/437) of residues occupying allowed regions (Figure 4).

Figure 3: 3D structure of refined multi-epitope CHIKV vaccine

Figure 4: Ramachandran plots profile of CHIKV-MEV 3D structure before and after refinement
Molecular docking with TLR-1/2
Determination of the interaction between vaccine constructs and immunological receptors (TLR 1/2) was predicted through molecular docking on the Cluspro 2.0 server program with an online protein-protein base. Molecular docking provides the possibility of predicting multiple protein interaction modeling with each other so that inspection can be done in parallel at the same time. A protein with a median score of -971.9 kcal/mol was used, as well as the lowest energy of -1079.0 kcal/mol (Figure 5).
Based on the molecular docking results between Chain B and Chain C, the complex that emerged resulted in 17 hydrogen bonds, 2 salt bridges, and 167 unbound contacts (Table 4).
Immune response simulation
To identify the immune response to vaccine exposure, an immune system simulation was performed using the C-ImmSim server. The CHIKV-MEV dose was set at 10 µL, 100 steps of simulations, single vaccine injection without LPS, and adjuvant to enhance immune response. The host HLA was selected by default from the server, consisting of A MHC class I (A0101), B MHC class I (B0702), and DR MHC class II (DRB1_0101). The simulation outcomes demonstrated a sequential pattern of the immune response, starting with an elevation in the secretion of IgM+IgG, which was subsequently followed by an increase in IgM and IgG1+IgG2 levels (Figure 6A). The initial immune reaction observed is the production of IgM + IgG. The subsequent stages of the immune response, characterized by B cell activation after vaccination, represent the secondary and tertiary phases (Figure 6B). There is also activation and formation of CD4+ T cells after exposure (Figure 6C). Cytotoxic CD8+ T cells were also indicated to have increased proliferation and activation, indicating a high potential for efficacy in the body (Figure 6D). Furthermore, cytokine and interleukin concentrations were at a stage where the immune response had detected the presence of the vaccine (Figure 6E).

Figure 5: Docking interaction complex between TLR-1/2 and CHIKV-MEV
Table 4: Hydrogen bonds between Chain B (TLR-1/2) and Chain C (Vaccine) of TLR 2-Vaccine complex


Figure 6: (A) Antigen and immunoglobulins. Antibodies are sub-divided per isotype, (B) B-lymphocyte population per entity-state (i.e. showing counts for active, presenting on class-II, internalized the Ag, duplicating and anergic, (C) CD4+ T-helper lymphocytes count sub-divided per entity-state (i.e. functional, resting, anergic, and duplicating), (D) CD8+ T-cytotoxic lymphocytes count per entity-state, and (E) The concentration of cytokines and interleukins. D in the inset plot is a danger signal

Figure 7: Population coverage prediction of CHIKV-MEV
Population coverage
The coverage population analysis was accomplished with an analysis tool provided by IEDB (Immune Epitope Database), resulting in candidate epitopes that have the highest frequency of occurrence in each coverage method. The data obtained from IEDB were analysed and it was found that there are variations related to MHC and HLA alleles in each geographical region around the world as a consequence of genetics and environment. Mapping population coverage in vaccine development is essential to consider for vaccine effectiveness. The results show that Europe has the highest combined MHC-I and MHC-II allele exposure with a percentage of 97.53%, while Central America has the lowest population coverage with a percentage of 2.77%. However, the global population coverage exhibits a remarkable outcome, indicating a 96.25% success rate (Figure 7).
Conclusion
Through an immunoinformatics study, a multi-epitope peptide-based vaccine targeting CHIKV has been developed. The prediction of epitopes, including linear B-lymphocyte (LBL), cytotoxic T-lymphocyte (CTL), and helper T-lymphocyte (HTL), was performed using the amino acid sequences of CHIKV E1 and E2 glycoproteins. The constructed vaccine has demonstrated the ability to elicit both humoral and adaptive immune responses by robust interactions with the TLR-1/2 receptor, as well as increased adaptive immune cell activation and the production of various cytokines capable of combating CHIKV. The vaccine design was also predicted to have high effectiveness in almost the entire global population. Despite containing comprehensive lymphocyte epitopes, the vaccine will be limited to detect CHIKV entities. Therefore, exploring potential epitopes from other immunogenic regions of CHIKV will enhance future chikungunya management. Moreover, thorough in vitro and in vivo studies with injection methods in animal models are holistically needed to validate the vaccine efficacy.
Acknowledgements
This research was funded by the Government of the Republic of Indonesia, Ministry of Technology, Research and Higher Education.
Disclosure Statement
No potential conflict of interest was reported by the authors.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Authors' Contributions
All authors contributed to data analysis, drafting, and revising of the paper and agreed to be responsible for all the aspects of this work.
ORCID
Nizam Zulfi Zakaria
https://orcid.org/0000-0002-3930-8286
Ahmad Fariduddin Aththar
https://orcid.org/0000-0003-1300-1961
Michelle Fai
https://orcid.org/0009-0009-6735-4368
Syeftyan Muhammad Ali Hamami
https://orcid.org/0000-0002-0443-520X
Viol Dhea Kharisma
https://orcid.org/0000-0001-9060-0429
Ahmad Affan Ali Murtadlo
https://orcid.org/0000-0002-7942-875X
Arif Nur Muhammad Ansori
https://orcid.org/0000-0002-1279-3904
Teguh Hari Sucipto
https://orcid.org/0000-0003-0512-2990
Rahadian Zainul
https://orcid.org/0000-0002-3740-3597
HOW TO CITE THIS ARTICLE
Nizam Zulfi Zakaria, Ahmad Fariduddin Aththar, Michelle Fai, Syeftyan Muhammad Ali Hamami, Viol Dhea Kharisma, Ahmad Affan Ali Murtadlo, Arif Nur Muhammad Ansori, Teguh Hari Sucipto*, Rahadian Zainul, A Novel Multi-Epitope Vaccine Design Targeting E1/E2 Envelope Glycoprotein of Chikungunya Virus: An Immunoinformatics Approach. J. Med. Chem. Sci., 2024, 7(2) 336-351.
)