Document Type : Original Article
Authors
Department of Pharmaceutical Chemistry, Faculty of Medicine and Health Sciences, SRM Institute of Science and Technology, SRM College of Pharmacy, Kattankulathur – 603203, Chengalpattu District, Tamil Nadu, India
Abstract
Thirty-six (36) new 5-oxo-imidazoline derivatives were studied using Insilco modeling against MCF-7 breast cancer cell lines. This research included QSAR, molecular docking, and pharmacokinetic analysis of the developed drugs. Based on the numerical evaluation of R2 = 0.7499, (R2adj) = 0.7061, (CCCext) = 0.6877, and (R2ext) = 0.6726, Model one performed best in the QSAR study. New derivative drugs with improved efficacy against estrogen-positive breast cancer (MCF-7 cell line) were designed using model number one. Derivatives of 5-(mercapto 1, 3, 4-oxadiazole)-5-oxo-imidazolines were predicted to be potent inhibitors of polo-like kinase 1 (plk1) based on molecular docking studies between the derivatives and the plk1 receptor. The results of the pharmacokinetic analysis of the new structures showed that they all met the criteria, including the Lipinski Rule of Five, and could move on to pre-clinical testing. They both showed that the MCF-7 cell line might be a unique therapeutic target in the fight against breast cancer.
Graphical Abstract
)
Keywords
- QSAR analysis 5-(mercapto 1
- 3
- 4-oxadiazole)-5-oxo-imidazolines Molecular docking studies Polo-like kinases (plk1) inhibitors Breast cancer
Main Subjects
Introduction
Uncontrolled cell multiplication, which affects the surrounding tissue and spreads throughout the body repeatedly, is how cancer is defined. That is a challenging problem [1]. Cancer is the most dreadful disease and a massive cause of human suffering and death, despite substantial advancements in high technology and social growth [2].
Breast cancer now accounts for one in eight cancer diagnoses and 2.3 million new cases in both sexes combined, replacing lung cancer as the most frequently diagnosed cancer worldwide [3]. According to estimates, 685,000 women have been died from breast cancer in 2020, accounting for 16% of all women's cancer deaths. The World Health Organization (WHO) recently launched the Global Breast Cancer Initiative in response to an earlier inadequate public health response to this development [4]. Patients diagnosed with breast cancer tend to share several characteristics. These include older age, failure to nurse for an adequate period, weight increase, early first pregnancy, inactivity, etc. [5]. Because of an extensive investigation into the processes and pathways by which cancer spreads and the discovery of various anti-cancer drugs, a breakthrough in cancer treatment extensive research into the techniques and ways by which cancer spreads and the discovery of different anti-cancer drugs, a breakthrough has been made in cancer treatment [2].
The progesterone receptor (PR) and oestrogen receptor (ER) positive luminal type of breast cancer (ER) is caused by overexpression of the oestrogen receptor (ER). This group includes about 70% of women with breast cancer identified as ER-positive (ER+). The MCF-7 cell line grows due to estrogens' ongoing ER activation [6]. Polo-like kinase 1 (Plk1) is a master regulator of the mitotic controller process and a popular therapeutic target. Its overexpression in the tumor is usually associated with a poor prognosis for the patient, and it is overexpressed in many different types of cancer [7]. In cancer cells, polo-like kinases (Plk1) promote aggressive proliferation and strong induction of cell-circle formation [8].
Due to genetic ambiguity and subsequent encouragement of mammalian cell modification, overexpression of Plk1 permits cells to circumvent hurdles. The most effective receptor for treating breast cancer has been identified as plk1. In human breast cancer cells, the estrogen receptor (ER), mediated by Plk1, controls gene overexpression. In a recent study, researchers observed that a group of 39 different imidazolone derivatives combined with a chalcone moiety suppressed the proliferation of MCF-7 breast cancer cells [9]. In natural products and artificial compounds, imidazole and its derivatives play an important role because imidazole scaffolds act as a bridge between synthetic organic chemistry and medicinal chemistry as they possess a plethora of biological applications like anticancer, antifungal, antiviral, anti-tubercular, anti-diabetic, anticonvulsant, antiulcer, and urease inhibitory. Since the imidazole molecules are extremely electron-rich and may readily bind to various enzymes and receptors, they exhibit a wide range of antiproliferative activity [10]. Angiotensin II receptor antagonistic activity, anticancer activity, antibacterial activity, cardio-activity, and other features have been demonstrated for compounds containing an imidazolone moiety [11].
Chemotherapy remains a prominent and efficient therapeutic approach. However, its efficacy is sometimes hindered by unfavorable toxicities, including weight loss, exhaustion, nausea, and appetite suppression. Consequently, the search for improved pharmacological alternatives with lower toxicity is imperative in order to effectively combat this condition [12]. The utilization of computer-aided drug design methodology has the advantage of time efficiency and greater efficacy in the development of therapeutic candidates. The objective of this study is to investigate the newly developed imidazolone derivatives through the construction of a quantitative structure-activity relationship (QSAR) model. This model was used to predict the anti-proliferative activities of the compounds. Furthermore, molecular docking studies were done using the derivatives and the plk1 receptor to gain insights into their interactions. The ultimate goal is to contribute to the discovery of anti-breast cancer drugs that are both less toxic and more effective in the treatment of breast cancer.
Materials and Methods
Computer applications
The software package for DS Visualizer 2021 consists of Chem Draw version 12.0.2, CHEMOPY descriptors, Conversion molecular formats open babel, molecular optimisation using Avogadro, and Molecular binding affinity using Auto dock version 4.2.1.
QSAR analysis
Dataset
In the works of Abo-Elanwar et al. (2019), 39 derivatives of imidazolones linked to chalcone moiety with anti-proliferative effects (IC50) on MCF-7 cancer cell lines were discovered. The anti-proliferative activity's inhibitory concentration (IC50) was calculated, and the logarithm scale (pIC50) was applied. Table 1 presents the pIC50 and the tabular version of the IC50, expressed in quantities of micro molar (Mm) units.
pIC50 =log10 (IC50 x 106).
Preparation of Molecular Structures and Optimization of 3D Geometry
The molecular structures were made with Chem Draw Professional 16 and converted into the mol2 format with Open Babel v2.4.1.17 (N.M. O'Boyle et al., 2011). To make the molecules' shape when hydrogens were added as optimal as possible, Avogadro V1.2.018 was also used. The molecular modeling force field (MMFF94) was developed using molecular mechanics in 1994 and was combined with the steepest descent method. The Avogadro tool and the "scoring function energy" have been used for each molecule to discover the ideal conformer with the lowest global energy. The same conformer was used for the investigation [13, 14].
Molecular identifiers
Table 1: Compounds in the dataset with structure, code, and experimental pIC50 value for polo-like kinase 1 inhibitory activity
.jpg)
.jpg)
.jpg)
The molecular descriptors for the 39 Imidazolone derivatives were optimized and calculated using CHEMOPY Server.
Pre-treatment and division of data set
Using Data Pre-treatment software GUI 1.2, the CHEMOPY Server results were processed to eliminate constant values and undesired descriptors [15]. To create a mathematical equation, the Kennard-Stone approach separated the derivatives into 12 validation sets and 27 calibration sets [16].
QSAR formula
A model was built using the Lack of Fitness Function (LOF) procedure and the QSRINS software. Anti-proliferative activities (pIC50) are the dependent variable, and model parameters are the independent variable. An external data set that has yet to be used during the model-development process should be used to test the validity of the built model [17].
Model validation
Models developed using "QSARINS" were evaluated for applicability and internally and externally validated. Selected models underwent the use of external validation the Q2 F1, Q2 F2, and Q2 F3 techniques, concordance correlation coefficient (CCC), internal validation using the Cross-validation leave-one-out (Q2LOO), Cross-validation leave-many-out (Q2LMO), Root mean squared error (RMSE), and Y-scrambling techniques. During the 5000 Q2LMO runs, 50% of the training set's objects are removed. Y-scrambling aims to eliminate chance association in the initial model by rearranging answer data across 5000 iterations. It is crucial to remember that the R 2 and Q2LOO should be more significant than the more fantastic scrambled ones and that the model's RMSE under prediction should have a lower RMSE. Since it measures the consistency between two variables, the CCC (CCCext) is a tool used to analyze the repeatability of models. Using leverage analysis, the descriptors employed and evaluated establish the theoretical area modeling’s applicability domain.
Stepwise regression, fitting with successive additions of descriptors, significantly improved the fit when the model could no longer be improved, and then the descriptors that had the most negligible statistically significant impact on model fit were dropped [18]. The process was repeated until no more descriptors could be deleted without suffering a statistically significant fit loss. Three models were made using these criteria. With the fewest descriptors, the highest (R2) was found with no descriptor co-linearity and the best prediction model was selected for future development, as provided in Table 2. The models' validity was evaluated using the "leave-one-out" (LOO) cross-validation technique. During the validation step, the model function is trained using data from every molecule but one [19]. The pIC50 for the chemical that was not a part of the study was then predicted using the model's properties. They utilize statistical measures like Q2 (cross-validated correlation coefficient), R2 (regression coefficient), SD (standard deviation), and SE (standard error) in the proposed QSAR models [20].
The least necessary values for evaluating the mathematical equation are listed in Table 2. The table parameters adjusted the derived equations' effectiveness and predictive capability [21].
ADME and drug-likeness predictions
To ascertain their pharmacological similarity, the best-fitting ligands obtained by molecular docking were put through ADME analysis. The ADME forecasts were created using a web application called Swiss ADME. A few characteristics (HIA) were Lipophilicity LogP, molar solubility in water LogS, BBB permeability, skin permeation, and human gastrointestinal absorption. The synthetic accessibility was predicted using a score from 1 to 10, where one indicates that the synthetic route is fairly pretty forward and ten indicates that the substance's structure is complex and that it is challenging to synthesize, along with such as Lipinski's rule of five, which have drug-like qualities. In addition, the bioavailability score was computed. Using the toxicity prediction, the possible toxicity of the inhibitors was estimated [22].
On Chemdraw Ultra 16.0, the recommended compounds' 2D structures were created to analyze their pharmacokinetic characteristics. When each structure was imported, the structural grin was entered into the (http://swissadme.ch/) interface. The ADMET features and criteria were produced by the Swiss ADME drug design study [23].
Molecular docking
The receptor Polo-like kinase 1(PKL1), in combination with BI2536, was used in molecular docking experiments on five drugs with significant pIC50. The ligand (compounds) were also converted to PBD format as indicated in Figure 1 (Receptor and Ligand 3D structures and the receptor was retrieved from Protein Data Bank (Code: 2RKU) and set using Discovery Studio software. To determine the binding affinity of the ligand and receptor, Autodock version 4.2.6 was used [24].
Results and Discussion
QSAR model developed and designed structures
Polo-like kinase (plk1) inhibitor compounds 1-39 were discovered using the pharmacological data of 39 5-oxo-Imidazoline derivatives from the literature (Table 1). As a measure of physiological activity, pIC50 (logIC50) was employed, and IC50 values represent the concentration of a chemical required to inhibit 30% of the tested in identical experimental conditions. Using principal component regression (PCR) and partial least square regression (PLS), it was determined whether there was a linear relationship between the IC50 and the relevant descriptors. The best prediction model, however, was produced by MLR, which included the fewest predictive elements and a high regression coefficient (R2) of the most crucial variables.

Figure 1: 3D Representation of prepared ligand-receptor
Model 01 was developed using MLR, and the descriptors had a standard error (SE) of 0.374 and accounted for 57% of the variation in biological activity.
Model 1
pIC50 = -3.4611 +2.6571*(Hato) +3.1781*(bcutp14) +1.5499*(MORSEC1)+-0.0429* (RDFU14)
ntr = 39 npred = R2=0.7429 R2adj= 0.7061 R2-R2adj = 0.0367 LOF = 0.0541Kxx = 0.3885 DeltaK = 0.0714 RMSEtr = 0.1762 MAEtr = 0.1433RSStr = 1.0241 CCCtr = 0.8525 s = 0.1912 F =20.2223Q2loo = 0.3558 R2-Q2loo = 0.1101 RMSEcv = 0.2105 MAEcv = 0.1702PRESScv = 1.4625 CCCcv = 0.7954 Q2LMO = 0.6076 R2Yscr = 0.1289 Q2Yscr = -0.2219 RMSEAVYscr = 0.3239RMSEext =0.3697 MAEext = 0.3167 PRESSext = 0.8199 R2ext = 0.6726 Q2-F1 = -0.2977 Q2-F2 = -0.3627 Q2-F3 = -0.1322 CCCext = 0.6877r2m aver = 0.2370 r2m delta = 0.4976.
Model 1 fits well and yields positive internal validation findings. With no outliers in William's figure, the external validation parameter values have somewhat decreased from the prior model. The descriptor correlation matrix for Model 1 is presented in Table 2. Figure 2 shows a scatter plot of the inhibitory actions of 5-oxo-imidazolines on polo-like kinase1, with expected values roughly matching the actual values. The final model's Kxy (inter-correlation among descriptors and response) versus Q2 LMO is depicted in Figure 3, and it demonstrates that the model is robust and stable because the "Leave many out" parameter values were near the model parameters. The structure-response relationship is broken, as the correlation coefficients of the final model are much higher than those obtained after endpoint scrambling Figure 4, which shows a Y-scramble plot of Kxy versus R2Yscr and Q2Yscr. In Figure 5, the forecast and the model's applicability range were illustrated using William's plot of standardized residuals vs. leverage levels. According to William's figure, all the compounds have leverage values lower than the warning h* of 0.462. They are in the model's application domain Figure 6. The importance of Q2F1, Q2F2, and Q2F3 are almost identical and of higher value as the CCC (concordance correlation coefficient) parameter values are raised. These results suggest that the best model was not found by accident and that the 5-oxo-imidazolines structure and polo-like kinase1 inhibitory activity are related.
In QSAR/QSPR studies, the 3D-MoRSE method was frequently applied, which creates representations of three-dimensional molecular structures using electron diffraction descriptors. Interatomic distances, scattering parameters (0–31 integer values), weighting by atomic properties like atomic charges (RDFC24), total surface area (PSA), MOE descriptors (EstateVSA5), and nuclear polarizability (MoRSEP3), as well as Moran autocorrelation, log 5 weighted by atomic polarizabilities (MATSP5), are some of the sources from which these data are derived. Compounds can be made more sensitive to the presence of particular molecular fragments using weighted descriptors.
In other words, the partial atomic charge can be used to calculate the distance between atoms having a high or low electron density. As the scattering parameters rise, the 3D-MoRSEC1 descriptors weighted with hydrogen-lighter schemes ought to exhibit less variation. Atomic mass, Van der Waals volume, and polarizability weightings have the least significant relative change in comparison, according to the literature. Distinct interatomic lengths may have distinct impacts, as shown by the dynamics of the cumulative sum of 3D-MoRSEC1 terms categorized by interatomic distance.
Table 2: The best model's correlation matrix


Figure 2: Scatter diagram of dataset compounds based on testing results of the best model equation

Figure 3: Visualization of dataset compounds using LMO scatter plots based on experimental values from the best model equation

Figure 4: Layout with a Y-scramble

Figure 5: The best model plot by William (h*, 0.455) warning value

Figure 6: Applicability domain plot of William's best model
Understanding the optimal interatomic distance in active compounds and comparing it to non-active compounds may help interpret 3D-MoRSEC1 descriptors by identifying the appropriate range of MoRSEC1 values required for the best activity, and thus aid in the interpretation of 3D-MoRSEC1 descriptors [25].
The connection between each descriptor in Model 1 and the activity is positive. The final model was used to calculate the pIC50 values for each drug, which are displayed in Table 3.
SAR of original data
Electron-withdrawing groups are substituted on the R1 position of the phenyl ring to improve the inhibitory activity of polo-like kinase 1, and based on the SAR analysis conducted on the original dataset utilizing molecular descriptors, it was seen that the incorporation of 5-mercapto-1,3,4 oxadiazole rings fused with a 5-oxo-imidazoline ring system resulted in a notable decrease in movement. In contrast, substituting a bulky and lipophilic group at the R1 position increased the activity. Hydrophilic substituents like methoxy and hydroxyl at the R1 position of the 5-oxo-imidazoline ring have been shown to improve polo-like kinase 1 activity (Scheme 1). The 5-oxo-imidazoline R2 position was substituted with various heterocyclic rings showed good activity Table 4. The analysis of the pki value using the model equation using molecular descriptors optimized compounds from the Chemopy server is shown in Table 5. Furthermore, molecular docking tests were carried out using combinations with greater pki values.
ADME Properties
It was discovered that ADMET disclosed a number of the physical and chemical characteristics of these novel molecules. These consist of additional factors, qualities, and the five principles of molecular synthesis (iLOGP). Number of heavy aromatic atoms (nAH), molecular weight (MW), rotatable bonds (RB), number of hydrogen bond donors and acceptors (HBD), octanol/water partition coefficient (iLOGP), molecular refractivity (MR), topological polar surface area (TPSA), and iLOGP. As demonstrated in Table 6, none of the synthesized compounds violates Lipinski's rule of five or the notion of drug-like properties more than once.
Table 3: The Best model equation predicted the PIC50 values of the original dataset


Scheme 1: SAR of designed novel 5-Oxo
Table 4: Designed novel 5-oxo-Imidazolinone compounds

Table 5: 2D structure, descriptor values, and predicted activity pIC50 for newly developed compounds
.jpg)
.jpg)
Table 6: ADME properties of designed the compounds

Discussion of the QSAR Model Generated with Imidazolone Derivatives
The results of the QSAR analysis demonstrated that the lowest possible estimates for the variables in the equation, as indicated in Table 2, were in agreement with the model's internal consistency and its external applicability. Using the validation compounds' model parameters shown in Tables 3 and 4, model 1 external validation was accomplished. The validity compounds dependability, and the calculated pIC50 of the calibration compounds were used to gauge the equation's efficacy.
Table 5 displays the biological, calculated, and residual values of compounds that are novel 5-oxo-imidazoline derivatives. The biological and calculated activities differ resulting in low residual values, demonstrating the high predicted Model 1. It is confirmed to be highly effective, robust, and significantly predictive by both internal and external validation. The definition of the model 1 parameter is shown in Table 6. The average effect of the model parameters reveals that (Hato, bcutp14, MoRSEC1, and RDFU14) carry positive coefficients, indicating bioactivities of the compounds would improve with a rise in those variables, while (RDFU14), which brings a negative coefficient, demonstrates that when the descriptor is lowered, the amount of experimentation performed by compounds derived from our novel 5-oxo-imidazoline derivatives. The statistical analysis indicates that the model parameters are not collinear, resulting in a robust equation, as shown in Table 6.
The pIC50 correlates well with the observed biological activity, as seen in Table 3, according to the graph of calculated activities (pIC50) against biological activities (IC50) displayed in Figure 1. Figure 2 shows that the values of both the calibration and validation compounds are distributed evenly across both sides of the graph, indicating that there are no systematic inaccuracies between the standardized residual versus bio-activities (Experimental activity). Figure 3 displays William's graph (standardized residuals vs. leverages), which reveals that all of the molecules were located inside the calculated warning leverage area of (h* = 0.56).

Scheme 2: Structures of best compounds on molecular docking studies
Table 7: Molecular docking interaction in some complexes
.jpg)
.jpg)

Figure 7: 2D and 3D representation of complex 2e
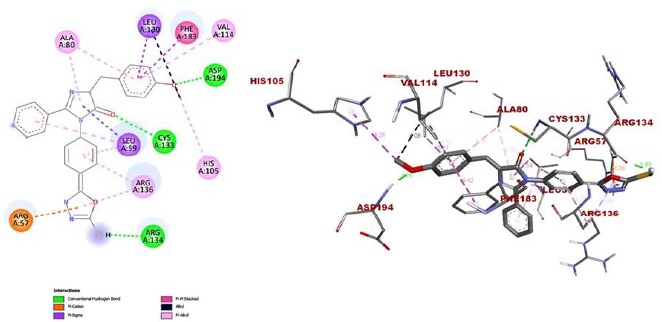
Figure 8: 2D and 3D representation of complex 2f

Figure 9: 2D and 3D representation of complex 2d
Analysis of molecular docking
Docking analysis was run on compounds containing derivatives of novel 5-oxo-imidazolines with the protein target Polo-like kinase 1 (PKL1) in complex with B12356. For these studies, seven compounds with high pIC50 were chosen, and of those 7, compounds 2e, 2f and 2d had the highest docking scores, with values of 11.33, 11.12 and 10.95 kcal/mol, respectively (see Table 7).
Compound 2e displayed backbone conventional hydrogen bonding interactions with the imidazolidinone group with CYS 133 (2.37A0), 5-mercapto-1,3,4 oxadiazole group with HIS 105 (2.11A0), and GLU 101 (1.86 A0). It also exhibited electrostatic pi-orbital cation, pi-donor hydrogen bond interactions with LYS 82 (2.50A0), and hydrophobic pi-sigma interactions with amino acids. Moreover, the substance established a Pi-Alkyl hydrophobic connection with two LEU 130 amino acids at a distance of 5.46A0, 4.95A0, two amino acids of LEU 59 at a distance of 4.83A0, 5.11A0, and also Pi-Alkyl hydrophobic interactions with ALA 80 (4.31A0), LYS 82 (5.20A0), CYS 67 (4.15A0), LEU 132 (5.02A0), ARG 134 (4.86A0), and ARG 136 (4.66A0).
Another instance of backbone conventional hydrogen bonding contact between the imidazolidinone group and CYS 133(2.20A0) was seen in compound 2f, 5- mercapto-1,3,4 oxadiazole group with ARG 134 (1.93A0) and 4-methoxy benzylidene group with ASP 194 (1.75A0), which is also having an electrostatic pi-orbital cation interaction with ARG 57 (4.08A0), hydrophobic pi-sigma interaction with the amino acid LEU 59 (3.59A0) and LUE 90 (3.31A0), Pi-Pi stacked hydrophobic interaction with PHE183 (4.42A0). Delocalized electrons in the pi-orbital of the benzene ring produced hydrophobic interactions with the alkyl groups of two amino acids of ALA 80 at a distance of (4.27A0), (5.15A0), LEU 59 (4.25A0), (5.13A0), ARG 136 (4.21A0), (4.48A0), HIS 105 (4.25A0), and VLA 114 (4.72A0) in the molecule.
Compound 2d showed four conventional hydrogen bonding interactions between the imidazolidinone group with CYS 133 (2.46A0), 5- mercapto-1,3,4 oxadiazole group with HIS 105 (2.22A0), and GLU 101 (1.87 A0) which is having an electrostatic pi-orbital cation and pi-donor hydrogen bond interaction with LYS 82 (2.53A0). Hydrophobic pi-sigma interaction with the amino acid LEU 59 (3.51A0), Pi-Pi stacked hydrophobic interaction with PHE183 (5.60A0), HIS 105 (4.06A0) possess Pi-sulfur interaction, and ten amino acids demonstrated Pi-alkyl hydrophobic interaction. Furthermore, the compound formed a Pi-Alkyl hydrophobic bond with two amino acids of ALA 80 at a distance of 4.41A0, 4.95A0, and then with two amino acids of LEU 59 at a distance of 4.83A0, 5.11A0, and also Pi-alkyl hydrophobic interactions with CYS 67 (4.35A0), LYS 82 (5.20A0), LEU 130 (4.97A0), LEU 132 (5.19A0), ARG 134 (4.98A0), and ARG 136 (4.58A0). Both the hydrogen bond and the hydrophobic interactions in the complexes showed that ligands 2e, 2f and 2d of 4-(5-mercapto-1, 3, 4-oxadiazolyl) 5-oxo-imidazoline derivatives are most active against Polo-like kinase 1(PKL1) in complex with B12356, respectively.
Conclusion
In this QSAR study, the original dataset was represented using descriptors, which resulted in a robust, stable, and predictive model. The establishment of a good statistical model has opened up a new avenue for studying descriptors in the suppression of Polo-like kinase 1 (PKL1). Internally and externally, the model is validated using software statistical parameters. To predict bioactivity, the model is tested on designed substances. To aid our QSAR model, we performed molecular docking studies on the best-predicted active chemicals to obtain structural and interaction information. Compounds 2e, 2f, and 2d obtained the highest docking scores, with scores of -11.33, -11.12, and -10.95 kcal/mol, to design and manufacture innovative estrogen-positive (MCF-7 cell line) breast cancer treatments, these compounds would serve as the most potent (PKL1) inhibitors, demonstrating a medical revolution. The pharmacokinetics analysis (drug-likeness test) performed on the designed molecules also showed that all compounds can advance to the next pre-clinical trial stage because they passed the drug-friendliness examination (ADME and other physicochemical properties) and because they complied with the Rule of Five, a benchmark used to evaluate the drug-likeness of compounds. The use of the MCF-7 cell line represents a significant step forward in the search for a permanent cure for breast cancer.
Acknowledgements
The authors would like to thank the Management of SRM College of Pharmacy for their support and encouragement and Dr. Paolo Gramatica, QSAR Research Unit, Insubria University, Italy, for the academic licensing software.
Conflict of interest
The Authors declared no conflict of interest in this article.
Authors' Contributions
All authors contributed to data analysis, drafting, and revising of the paper and agreed to be responsible for all the aspects of this work.
Funding
No funding was received to assist with the preparation of this article.
Ethical approval
There is no ethical approval in this article.
ORCID
Yellasubbaiah N
https://orcid.org/0000-0001-7109-6992
Velmurugan V
https://orcid.org/0000-0001-5316-6854
HOW TO CITE THIS ARTICLE
Yellasubbaiah N, Velmurugan V. QSAR Modeling, Molecular Docking, and ADME Studies of Novel 5-Oxo- Imidazoline Derivatives as Anti-Breast Cancer Drug Compounds against MCF-7 Cell Line. J. Med. Chem. Sci., 2023, 6(12) 3087-3112.
)