Document Type : Original Article
Authors
SRM College of Pharmacy, SRMIST, Kattankulathur, Chennai, Tamil Nadu-603203, India
Abstract
Dipeptidyl peptidase 4 (DPP-4) inhibition is a promising therapy for type 2 diabetes. Inhibition of DPP-4 limits the breakdown of glucagon-like peptide 1 (GLP-1), hence increasing functional GLP-1 levels. This boosts the secretion of insulin and decreases glucagon release, resulting in a reduction in blood sugar levels. In an effort to discover the chemical and structural prerequisite for DPP-4 inhibition, computational investigations involving Quantitative Structural–Activity Relationship (QSAR) studies were performed on 63 compounds with pIC50 values ranging from 7.0 to 9.744. With a good R2 (0.716) and cross-validated correlation coefficient value Q2 LOO (0.6120), a model was created that quantitatively describes the relationship. The regression approach systematically gives that the topological state of an atom, the presence of CF3 at the second position of the triazole ring (knotpv), the polarizabilities (RDF15p), the atomic masses (MATS3M), the heteroatom, and the valence distribution had a significant effect on DPP-4 inhibition (Chiv4pc). In addition, docking results revealed favorable contacts between triazolopiperazine analogues and catalytically significant amino acid residues, including HIS740, SER630, ASN710, and GLU205. Comparing the interactions of the most active compound 4 to those of the standard Sitagliptin reveals comparable binding energies. The study demonstrates that substituting CF3 at the second position of the triazole nucleus and incorporating polarity-altering groups are advantageous for inhibiting the DPP-4 enzyme.
Graphical Abstract
)
Keywords
Main Subjects
Introduction
In 2017, 462 million people worldwide were diagnosed with type 2 diabetes mellitus (T2DM), accounting for 6.28 percent of the global population and 6059 cases per 100,000. Diabetes is the tenth leading cause of mortality, accounting for around 1 million fatalities yearly, with many more deaths occurring later in life. By 2030, the global prevalence of type 2 diabetes is anticipated to reach 7079 individuals per million, with rates increasing in every country [1]. Type-2 diabetes (T2DM) is perhaps the most common type. It is a chronic condition characterized by increased blood glucose levels and insulin resistance. DPP-4, a serine protease found in the blood, controls glucose metabolism by degrading the incretins glucagon-like peptide 1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP) produced after a meal.
By inhibiting DPP-4, the amounts of GLP-1 and GIP are increased, which suppresses glucagon release and enhances insulin production, ultimately lowering blood sugar levels [2]. DPP-4 is a ubiquitous physiological enzyme that is soluble in blood and membrane-anchored in tissues. Inhibiting DPP-4 activity makes sense in type 2 diabetes since it decreases peptide cleavage and hence enhances endogenous incretin hormone activity [3]. Although strict glycaemic control decreases morbidity and mortality associated with type 2 diabetes is difficult to maintain and often unsuccessful.
Current anti-diabetic medications exhibit a progressive loss of effectiveness, poor tolerability, and low compliance owing to various side effects, including severe hypoglycaemia, weight gain, oedema, nausea, and gastrointestinal disturbances. Thus, new ways were required to maintain glycaemic control while avoiding hypoglycaemia and other adverse consequences [4]. As a result of recent advancements in this sector, new antidiabetics such as incretin mimics, amylin analogues, GLP analogues, dipeptidyl peptidase-4 (DPP-4) inhibitors, PPAR agonists, and PPAR antagonists have been developed [5]. Using DPP-4 inhibitors to treat type 2 diabetes is a novel and intriguing technique. "Gliptins" or "DPP-4 inhibitors" are weight-neutral, have a lower risk of hypoglycemia, as well as provide long-term post-meal glycemic management [6].
Between 2006 and the present, substantial research efforts have resulted in the introduction of a handful of DPP-4 inhibitors with somewhat identical activity, including sitagliptin [7], saxagliptin [8], vildagliptin [9], linagliptin [10], alogliptin [11], and omarigliptin [12], to name a few examples. Identifying the binding interactions between both the DPP-4 enzyme and its inhibitors is critical for developing potential DPP-4 inhibitors. According to the previously described structure-activity relationship (SAR) of DPP-4 inhibitors developed from sitagliptin, the DPP-4 binding site is made up of many sub-sites, comprising S1, S2, and huge S2 pockets [13]. S1 and S2 contain Arg125, Ser209, Phe357, Arg358, Tyr547, Ser631, Val656, Trp659, Tyr62, Tyr666, Asn710, and Val711, whereas S3 has Asn281, Leu294, Leu340, Val341, Ala342, and Arg343, which is an alternative binding site and is regarded as an allosteric binding site [14].
It is believed that hydrophobic amino acids at the enzyme's N-terminus (Glu205, Glu206, and Tyr662) promote substrate selectivity [15]. Many biologically active medications have been discovered with the use of computer-aided drug design, specifically protein-ligand docking [16] For a few reasons, figuring out how ligands, which are typically tiny organic molecules, attach to their large protein targets is crucial for both studying biological mechanisms and developing effective therapeutics [17]. Nevertheless, a drug's polarizability is essential in characterizing the structural composition of a medicine. Thermodynamics and orientation. Additionally, it has benefits in Pharmaceuticals to produce Quantitative Associated Structural Activities (QSARs) and drug development [18].
In order to better understand how medications and nutritional supplements work in the human body, many scientists have turned to computational approaches, including molecular docking, virtual screening, and qualitative-structural activity relationship [19]. Factors that increase the likelihood of acquiring type 2 diabetes include age 45 and up, the presence of the metabolic syndrome, high fasting blood glucose, a higher body mass index (BMI), and a glycosylated hemoglobin (HbA1c) value that is greater than 6% [20]. Insulin resistance precedes type 2 diabetes mellitus (T2DM) and leads to the failure of -cell function and, thus, a gradual decline in insulin production due to both hereditary and environmental factors, including inactivity and obesity [21].
The piperazine analogue functions as an irreversible mechanism and serves as a lead for the majority of dipeptidyl peptidase-4 inhibitors. Nonetheless, Piperazine compounds show potent anti-diabetic properties. Therefore, researchers have made ongoing efforts to synthesize Piperazine derivatives as DPP-4 inhibitors. Sitagliptin, a beta-amino acid-derived inhibitor with additional changes to the 8th position of the triazolopiperazine nucleus by the inclusion of a benzyl, modified benzyl, or pyridyl group, exhibited effective inhibition with an IC50 value of 0.32nM (measured against mouse plasma) [22]. In continuation of our computational chemistry-based drug discovery strategy, we discuss the molecular modelling studies performed on Triazolopiperazine analogues with an effort for to optimization the lead compound. Using QSAR and docking analyses, the main structural characteristics of Triazolopiperazine analogues required for the inhibition of dipeptidyl peptidase-4 were identified (Figure 1).

Materials and Methods
Quantitative structure-activity relationship (QSAR) program
The quantitative structure-activity relationship (2D) investigations has have been carried out with the QSAR-INSURBIA software QSARINS (2.2.3, Insubria and Varese Universities, Italy) [23]. QSAR-INSURBIA software uses GA and MLR to build highly predictive and easy-to-interpret QSAR models. The molecular modelling simulation program Auto Dock Tools 1.5.7, Institution for Advanced Study at Scripps in San Diego, California, was used for docking research [http://www.autodock.edu] [24]. ChemDraw Professional 15.0 was used to design the structures [25] and cleaned by the option clean structure found in the toolbox followed by energy minimization by using chimera version 1.16, by generating smiles for the structures, and minimizing the energy [26]. Protein-ligand interactions were carried out using the BIOVIA Discovery Studio 2021 [27].
Model development by QSAR
Minimization of energy as well as dataset splitting
From the literature, a variety of Triazolo piperazine compounds assessed against human plasma were chosen (Table 1). Chemdraw 15.0 version was utilized to draw the chemical structures and converted to mol2 format using OpenBabel 2.4.1 version, an online chemical toolbox for the conversion of structures into various formats. Then, the structures were optimized using Chimera version 1.16 software for energy minimization. The minimized structures served as inputs for descriptor calculation. The DPP-4 inhibition in nanometres was logarithmically transformed to standardize experimental data values (PIC5014log 1/IC50), and the pIC50 ranged from 7.00 to 9.744. The test set was chosen using a randomized selection [23] procedure with an odd chance of around 20%. The dataset was divided in half, with 51 molecules in the training set and 12 molecules in the test set [28].
Calculation of descriptors
Molecular descriptor calculations were performed on the energy-minimized compounds using PaDEL free online software version 2.21 to calculate 2D descriptors [29]. To eliminate the correlations among many descriptors, the attributes which were screened beforehand by removing any values are lacking that do not include zero values after a sizable collection of descriptors had been generated. Moreover, descriptors with more than 0.30 values were filtered out using paired correlation. After excluding all the descriptors based on the correlation matrix, the topological 2D descriptors knotpv [30], Chiv4pc, RDFP15, and MATSM3 exhibited great co-relation with activity. The most contributing factors for the DPP-4 inhibition in triazolopiperazine nucleus are these descriptors. The knotpv descriptor indicates the difference of between two connectivity indices, P valence cluster 3 and P valence path/cluster 4. Both are proportional to the molecule's size and the number of HBD and HBA. The intermolecular mobility indices could be used to understand the molecular connectivity indices [31]. MATS3M, a Moron autocorrelation descriptor, contributes lag 3/weighted by atomic masses (2D autocorrelations) [32]. RDF15p, a radial distribution function – 015 / weighted by the atomic polarizabilities [33, 34]. Chiv4pc, a connectivity descriptor, and can be resembled as an isobutane skeleton has a simple chi valence path-cluster 4 index that is particularly sensitive to the proximity of skeletal branch points, and includes information about heteroatom and valence state [35]. After numerous trial models, the best models created 2D descriptors, which are shown here.
Validation of statistical model
A realistic evaluation of a QSAR model's true predictive capability must be carried out most rigorously and realistically feasible, as the real value of a QSAR model resides in its ability to reliably forecast the modelled property for novel chemical substances [36]. Multiple Linear Regression is still the most frequent and transparent method, where models are represented by clearly articulated mathematical equations. Despite the fact that Even though all QSAR models, both linear and nonlinear, are based on algorithms. The following are the five guiding concepts for evaluating the QSAR model: i) The model ought to be able to provide a specified endpoint in this context. The term "endpoint" refers to a pharmacological property or activity that might be tested and used in modelling. ii) A clear-cut and explicit algorithm is required for a QSAR model to be acceptable. iii) A Defined Applicability Domain, iv) acceptable goodness-of-fit, robustness, and predictability measures, v) a mechanical explanation, if possible. criterion were used to verify the model's stability [37, 38], i.e., leaving one out of the equation, producing predictions for the molecule that was left out after deleting a compound out from the dataset, and constructing the model with the remaining compounds.




Internal validating procedures such as the Q2 LOO In order for internal validation requirements to be met, the values of Q2 LOO and R2 must be comparable. Leaving many out (LMO) is a more powerful cross-validation strategy that is employed in validation criteria and tests the model's predictive abilities by excluding a lower fraction of compounds. If (R2 LMO and Q2 LMO) values were equivalent to R2 and Q2 LOO values, the model is termed stable and robust. The Y scrambling process in QSARINS was used for the external validation [39]. The predictability of the Y-response scrambled data is evaluated to determine if the model's correlation is due to chance. Due to the use of this approach, there is no discernible pattern to the distribution of the responses with little or no relationship to the descriptors, and the relationship between the model and the descriptors is examined. The R2 and Q2 values of each phase, as well as and their averages (R2 YS and Q2 YS), must be less than the model values if the internally accepted validation parameters are met. Validation criterion requiring a mechanistic hypothesis of DPP-4 inhibition by triazolopiperazine derivatives
Virtual molecular docking
Protein processing
The process of molecular docking is a high-tech computer strategy that can be used in a variety of various situations, which describes how a ligand fits into the active region of a protein, where interaction and stimulation occur [40]. The portion of a protein's active site made up of residues that form transient bonds well with the substrate and amino acids that accelerate the counteraction on the ligand is referred to here as an active site. The X-ray molecular structure of Dipeptidyl peptidase -4 (PDB ID: 5Y7K) was derived with a resolution of 2.51 A֯ employing X-ray crystallography. Data for the R-value of 0.241 came from the PDB repository (www.rcsb.org). The recommended value of crystallinity with a resolution of 2.0 or even less, and indeed the R-value is 0.2 or less; these values indicate the quality of the standard protein currently utilized.
Docking parameters
Autodock 1.5.7 [24] was used for docking the molecules. In order the grid map spacing of 60*60*60 points and the X= 98.6, Y= -22.02, and Z= 54.52 dimensions were used as the centre point to perform docking calculation. A total of 500 thousand energy evaluations were completed for each and every single of the docking study with. 100 distinct runs. Docking statistics are based on energy-scoring models with other docking settings set to default.
The binding values obtained were used to aggregate the output of all the derivatives, and the optimal position of docked ligands with the lowest energy conformations was recorded. Only the highest-scoring poses were considered in all docking studies. Sitagliptin was employed as a reference ligand to even further confirm the docking approach.
Discovery studio visualizer
The Discovery Studio Visualizer program was used to analyze and visualize the 2-dimensional and 3-dimensional ligand interactions. The software's receptor-ligand interactions module was utilized to assess the ligand interactions from among components [41].
Results and Discussion
Relationship between quantitative structure and activity
As a means of elucidating the connection between molecules (descriptors) and biological function, triazolopiperazine derivatives were selected and quantitatively investigated (pIC50). By developing output-defining models, the statistical results of the descriptor evaluation on the chemical structures were calculated and compared to the biological activity. This study aimed to establish a linear association between the descriptors that contribute the most to potency.
Physicochemical parameters, steric interactions, electronic and electro-topological data were combined to create a total of 1444 [23] structured descriptors (1D and 2D) using PaDEL. After excluding descriptors with zero or near-constant values and those with high pair-wise correlation (R>0.70), most preferable pharmacologically-relevant descriptors, were found. After rigorous refinement, 117 descriptors were chosen for the model development. Using the QSARINS program and MLR, a large number of QSAR models has have been developed utilizing various descriptor combinations. Validation of the best model yielded satisfactory Q2 and R2 values.
We used MLR regression analysis to create a model to explore the DPP-4 inhibition of triazolopiperazine derivatives in this research, 12 compounds (4, 6, 10, 23, 25, 33, 37, 43, 44, 51, 56, 63) were utilized as a prediction set to apply the best model, and the remaining compounds were grouped as training set compounds. A predicted and stable Multilinear regression model has greater R2 and equivalent Q2 LOO values, as well as a minimum error value, like SEE which means (standard error of the estimate), and indeed by a minimum quantity of descriptors. And the parameters were cut down to four descriptors based on a ratio of 4:1 to eliminate correlations caused by the excessive number of descriptors. The descriptor that most significantly increased potency for each of the four compounds was selected as the attribute.
Equation (1) the finest linear model with given statistical values is shown in the following.
Activity =6.2930+3.6587(knotpv) + (-1.1437*Chiv4pc) +0.1194(RDF15p) +4.2799(MATS3m)
Fitting criteria
n= 63, R2 = 0.7167, R2 adj= 0.6802, R2- R2 adj =0.0365, LOF= 0.2115, Kxx= 0.3968, Delta K= 0.0756, RMSEtr= 0.3577, MAEtr= 0.2808, RSStr= 4.6059, CCCtr= 0.8350, S= 0.3852, F= 20.0796.
Internal validation criteria
Q2LOO= 0.6120, R2- Q2LOO= 0.1047, RMSEcv= 0.4186, MAEcv= 0.3280, PRESScv = 6.3357, CCCcv=0.7808, Q2LMO = 0.5797, R2Yscr = 0.1150, Q2 Yscr = -0.2125, RMSE AV Yscr = 0.6316
External validity criterion
RMSEExt = 0.2161, MAEExt = 0.1632, PRESSExt = 0.3185, R2Ext = 0.9212, Q2F1 = 0.8939, Q2F2 = 0.8911, Q2F3 = 0.8966, CCCExt = 0.9480
The descriptors mentioned in Equation (1) are defined and shown in Table 2. With a high R2 (coefficient of determination) R2 = 0.7167 along with a greatest F value (Fitness value) F = 20.0796, this model is deemed the best, indicating that it complies with the internal validation requirements and excellent fitting criteria. The correlation analysis of the descriptors utilized throughout the investigation is displayed in Table 3, demonstrating that, in this case, there isn't any association between the descriptors employed. As shown in Figure 2, the testing action and predicted results show a linear connection in the scatter plot.
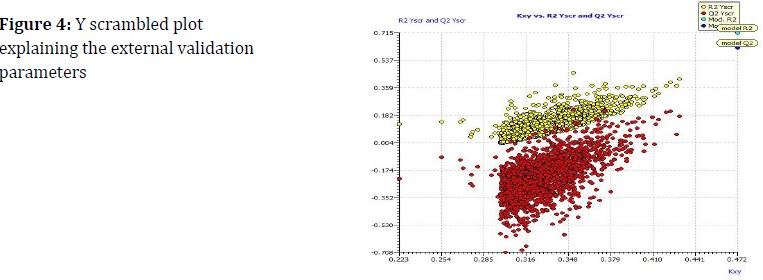
The association between x-descriptors and y-activity was depicted with Kxy vs. Q2LMO of the finalized model in Figure 3, indicating that the model is consistent and predictable because the LMO parameter values obtained were closest to the model parameters. A Y-scramble plot of Kxy vs. R2YSCR was used to evaluate the external validity (Figure 3), and Q2 Yscr reported a smaller value than the values in the model.

Figure 4 shows William's plot of residual values vs. leverage values, which demonstrated the applicable domain of the model. having leverage values below than the h* threshold of 0.417. Especially in comparison to the parameters of the CCC (concordance correlation coefficient) model, the values of Q2F1, Q2F2, and Q2F3 were virtually identical. The model has been verified both internally and externally as the consequence of a genuine association between the structural characteristics and DPP-4 inhibition, and not a mere coincidence.
The two key PaDEL descriptors for biological activity have been identified as Knotpv and MATS3m (a topological state and Moron auto co-relation descriptor). A topological descriptor is the one which gives the information about the molecular connectivity index and is responsible for intermolecular accessibility. The difference of between the two connectivity indices, P valence cluster 3 and P valence path/cluster 4, is described by the molecular descriptor knotpv. Both variables correlate favorably with the molecule's size and the number of HBD and HBA. The hydrogen bond acceptors are important in determining the permeability of triazolopiperazine derivatives through membranes. Intermolecular accessibility can be interpreted in terms of the molecular connectivity indices. MATS3m corresponded to the energy of the molecular orbital with the maximum occupation in the ground state, atomic masses, as well as electronegativities. Chiv4pc is a connectivity descriptor that encapsulates relevant information on a branch point, with an emphasizing on the adjacent branch, and specifies heteroatom and valence information. This descriptor favorably adds to the MLR model. The biological processes of DPP-4 inhibition can therefore be enhanced by raising the descriptor index of its segments.
Analysis of molecular docking
The best-fitting QSAR model's structural parameters were investigated further than just their required interaction with human Dipeptidyl peptidase IV (5Y7K). The crystalline structure of human DPP-4, as seen by an X-ray, has been retrieved, and the protein with amino acids was generated by eliminating crystalline the waters and co-crystallized ligands. In Autodock 1.5.7 protein preparation wizard, the hydrogen atoms were introduced to the molecule introducing Polar Hydrogens. These investigations uncovered probable potential connections with logical groups that attach about amino acid residues in DPP-4's catalytic site and pinpoint the pharmacophore requisite for binding purposes.
Molecular docking assisted us in discovering derivatives with better interaction energies (binding energies in kcal/mol) in order to explore the structural properties that do have the significant contributions to biological activity. The binding efficacy of a ligand to a protein is expressed as the binding energy per atom of the ligand to a protein. According to Table 3, the compound with the best binding energy was compound 4 (-10.59 kcal/mol), and the compound with the lowest binding energy was found in compound 32 (-3.95 kcal/mol) (Table 4).
Compounds 48 and 50 with significant experimental biological inhibition (pIC50) with 9.744 nM and 9.508 nM had binding energies of -7.06 and -7.27 kcal mol-1, correspondingly. The reference substance sitagliptin's binding energy was determined to be -7.66 kcal mol-1. Compounds 18, 49, 54, 57, 58, and 59 exhibited binding energies that, respectively, were close to our reference molecule at -7.31, -7.95, -7.58, -8.46, and -7.06 kcal mol-1.
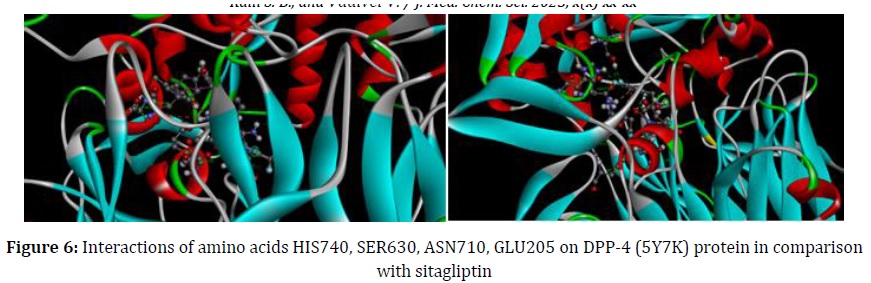
According to the same binding scores as the standard drug, the relevant interactions for compound 32 were ASP663, GLU205, TYR631, VAL656, HIS740, SER630, ASN710, GLU206, HIS126, PHE357, TYR662, VAL711, ARG125, and TYR666. Compared to the standard SITAGLIPTIN, the test compounds displayed comparable and noticeable interactions with the HIS740, SER630, ASN710, GLU206, HIS126, PHE357, TYR662, VAL711, and ASN710 (Figure 5).

Compound 50, with a pIC50 of 9.744 nM, was assessed for docking interactions with human DPP-4 as a potential candidate with the characteristics favorable for DPP-4 inhibition (5Y7K). Hydrogen bonds exist between residues such as ARG125, TYR666, HIS363, TRP305, and ALA306, according to Investigations into Docking Systems of triazolopiperazine derivatives for DPP-4 inhibition. Molecules with the highest binding energies, such as 50, form more hydrophobic interactions with DPP-4.
According to the docking data, triazolopiperazine derivatives bind to the DPP-4 active site with good interaction, much like sitagliptin does. The structures of the indicated standard inhibitor sitagliptin and compounds 4 and 5 docked into the active catalytic site of the DPP-4 enzyme are shown in (Figure 6).
Structural activity studies
Using the QSAR and docking data, a structural analysis of the series of triazolopiperazine analogues was performed. Taking into account the requirement of CF3 at the second position of the triazole ring compounds 48 (pIC50 = 9.508) and 50 (pIC50 = 9.744) matched with the best-fitted QSAR model. In comparison Compared to hydrogen, the CF3, which exhibits electronegativity and is likewise chosen as fluro, is smaller in size. CF3 enhances the binding affinity of the compound to the target protein, and its interaction influences the polarity of other groups involved in the compound, which refers to the topological descriptor in the QSAR model.

They demonstrated an inhibition range of 7.00 to 9.744 using a dataset of 63 compounds. Compounds 18 and 36, which exhibit the minimum experimental inhibition with pIC50 values of 7.00 and 7.040, identify the existence of hydrogen at the second position of the triazole ring and the absence of CF3. In compound 36, the activity was greatly reduced by the presence of a methyl group in the second position and a hydrogen atom in the third position of the piperazine ring.
Groups like phenyl, piperazine, and triazole demonstrated similar interactions from the docking action of ligands with the receptor. Better interactions have been seen with compound 4, which has a high binding energy of -10.59 kcal/mol. The introduction of another triazole ring at the second position of piperazine leaded led to decreased activity (compound 32). The presence of CF3 at the second position of the triazole ring and three fluro groups in the phenyl ring has shown better inhibition of the DPP-4 enzyme. With Consideration of the enumerated pharmacophores in the modelling of triazolopiperazine analogues would be a watershed moment in the DPP-4 inhibitor drug discovery and development.
Conclusion
The QSAR and docking analysis confirmed that the presence of the CF3 group at the second position of triazole is described by the knotpv topological molecular descriptor. The secondary amine (NH2) present acts as an electron-donating group or shares electrons’ lone pair to form a covalent bond with a biological target is described by the chiv4pc molecular descriptor. Docking further validated the pharmacophores’ presence, indicating favorable binding energies and comparable Protein-Ligand interactions. Hydrogen bonding interactions and molecular modelling studies discovered the critical contacts that coordinate with the essential catalytic interactions, such as ARG125, TYR666, HIS363, TRP305, ALA306, HIS740, PHE364, and THR304. The Insilco experiment results specify the structural requirements for subsequent ligand design that targets DPP-4 inhibitory action.
Acknowledgements
The authors are grateful for the support of the Research Council of SRMIST and the Dean of the SRM College of Pharmacy. Prof. Paolo Gramatica, University of Insurbia, Varese, Italy, is also acknowledged for making it accessible to the QSARINS software.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Authors' contributions
All authors contributed to data analysis, drafting, and revising of the paper and agreed to be responsible for all the aspects of this work.
Conflict of Interest
We have no conflicts of interest to disclose.
ORCID:
V Velmurugan
https://orcid.org/0000-0001-5316-6854
HOW TO CITE THIS ARTICLE
Swarna Bharathi Kalli , Velmurugan Vadivel. Qsar and Docking Studies of New Triazolopiperazine Derivatives as Potent Hypoglycemic Candidates, J. Med. Chem. Sci., 2023, 6(7) 1598-1613
)