Document Type : Original Article
Authors
- Deto Ursul Jean-Paul N’Guessan 1
- Songuigama Coulibaly 1
- Fulgence Kondo Kassi Kassi 2
- Pierre-Olivier Delaye 3
- Melanie Penichon 3
- Cécile Enguehard-Gueiffier 3
- Hassan Allouchi 3
- Mahama Ouattara 1
1 Department of Therapeutic Chemistry and Organic Chemistry, UFR Pharmaceutical and Biological Sciences, FHB University, 01 BP V34 Abidjan, Ivory Coast
2 Department of Parasitology-Mycology-Zoology, UFR Pharmaceutical and Biological Sciences, FHB University, 01 BP V34 Abidjan, Ivory Coast
3 University of Tours, Faculty of Pharmacy, EA 7502 SIMBA, 31 Avenue Monge, 37200, Tours, France
Abstract
The increase of immunodeficiency situations such as HIV and cancer is stemmed from the expansion of fungal infections due to the genus Candida. Although Candida albicans remains the most widespread species in pathogenic isolates, its epidemiological impact in human infectiology has declined in favor of new emerging species of Candida refractory to conventional treatment. Faced with this situation, we decided to contribute to the development of some imidazo [1,2-a] pyridinyl-arylacrylonitriles, as potential new anticandidosics. We proposed the design by molecular hybridization and the synthesis of some imidazo [1,2-a]pyridinyl-arylacrylonitriles following a Knoevenagel condensation reaction between aldehydes and various arylacetonitriles. Furthermore, we carried out the evaluation of the antifungal activities of these hybrid derivatives against Candida albicans, Candida tropicalis and Candida glabrata using the microplate dilution methodology. In the end, imidazo[1,2-a]pyridinyl-arylacrylonitriles turned out to be molecules with strong antifungal activities. The best anticandidosis profile on the three Candida species tested was obtained with the 3-chlorinated compound (5), the MICs of which varied between 357.5 - 0.52 µM. Likewise, the doubly modulated derivative (3c), was particularly illustrated by its good efficacy against Candida tropicalis. These two best compounds can be proposed as the "hit molecules" for further pharmacomodulations in order to have a drug candidate for anticandidosis purpose.
Graphical Abstract
![Synthesis and SAR of Imidazo[1,2-a] Pyridinyl-Phenylacrylonitrile Derivatives as Potent Anticandidosis Agents](javascript:loadModal('Synthesis and SAR of Imidazo[1,2-a] Pyridinyl-Phen ... ', 'data/jmcs/coversheet/671632726320.jpg'))
Keywords
Main Subjects
Introduction
In the past decades, invasive candidiasis has emerged as one of the most common fungal infections [1]. This widely disseminated and bloodstream infection often gives rise to high mortality rates globally, mostly in at-risk patients such as hospitalized, immunocompromised patients, or the elderly [2]. However, therapeutic options for the treatment of invasive candidiasis are currently limited to only three classes of antifungal agents: Echinocandins (caspofungin, micafungin or anidulafungin), azoles (fluconazole, voriconazole) and macrocyclic polyenes (amphotericin B, nystatin) [2]. In addition, there is an emergence of other clinically relevant species recovered from blood stream infections including C. tropicalis, C. dubliniensis, C. lusitaniae, and the most recent, i.e., C. auris [3]. These Candida species are a serious threat for public health because of their high likelihood of intrinsic and acquired resistance. In addition, drug treatment of these candidiasis has become a public health problem since the misuse of antifungals has contributed to the proliferation and emergence of drug- resistant strains of Candida. Therefore, treatment of fungal infections is becoming a significant concern to clinicians and there is a pressing need to discover new antifungal-drugs to overcome resistance of Candida sp.
Imidazo[1,2-a]pyridine, when attached to functional groups or other rings, leads to hybrid molecules exhibiting high pharmacological activity. Many of imidazo[1,2-a]pyridine derivatives such as soraprazan, divaplon, zolpidem, olprinone, and zolimidine are used in therapeutic for diverse diseases. In addition, synthetic hybrid compounds such as imidazo[1,2-a]pyridinyl-phenylpropenones, reported in a previous work [4], have shown remarkable antifungal activity with minimum inhibitory quantities of between 5 and 0.31 µg. Recently, we reported the discovery of phenylacrylonitriles with an imidazo[1,2-a]pyridine core having good anthelmintic activities against Haemonchus contortus [5].
In this study, we focused on the imidazo[1,2-a]pyridine ring, because it is a structural isostere of benzimidazole, the heterocyclic nucleus of chlormidazole, and the first antifungal of total synthesis[6]. Moreover, this scaffold is a fused imidazole which is the basic core of antifungal class of Azoles [6].
In addition, the acrylonitrile chain, is currently used as an interesting constituent for new nitrile-containing pharmaceuticals [7]. This promising functional group is present in the structure of the new topical antifungal azoles developed and marketed in Japan, luliconazole and lanoconazole (Figure 1) [7,8].
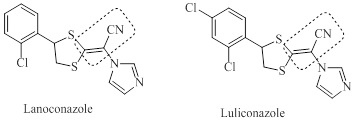
Figure 1: Structure of ravuconazole et luliconazole
Both drugs showed a dual interaction for inhibiting sterol 14R-methylase in fungi. Indeed, they act through a π-interaction between the enzyme and the chlorobenzene ring. In addition, because conjugation of the nitrile with an additional electron-withdrawing group facilitates Michael addition, the dithioalkenenitrile of ravuconazole and luliconazole acts as a Michael acceptor [7].
Thus, in view of the above mentioned properties of these chemical entities and by applying hybridization [9] of bioactive entities, we designed chemical profile of imidazo[1,2-a]pyridinyl-arylacrylonitriles (Figure 2).

Figure 2: Conception of imidazo[1,2-a]pyridinyl-β-phenylacrylonitriles as antifungal agents
Taking this into account, we carried out the following study to identify a molecular hit likely to be developed as an antifungal drug candidate. Accordingly, we reported herein the synthesis and the antifungal activities of these new acrylonitrile hybrids against clinical strains of Candida. Specifically, we focused on the determination of the Minimum Inhibitory Concentrations (MIC) of imidazo[1,2-a]pyridinyl-acrylonitriles or acrylamides against Candida albicans, Candida glabrata and Candida tropicalis. Then, SAR investigation was undertaken to discover new anticandidosis agents based on imidazo[1,2-a]pyridine.
Material and methods
General
Melting points were determined by using a Kofler bench and were uncorrected. NMR experiments were performed at 300 mHz (1H) and 75 mHz (13C) on a Bruker-Avance 300 MHz spectrometer using CDCl3, with reference to tetramethylsilane as an internal standard. Assignment of carbons noted C* may be interchanged. Mass spectra were recorded on a Hewlett Packard 5988A spectrometer or on a Shimadzu QP 2010 spectrometer by direct inlet at 70 eV. All reagents were used directly as obtained commercially. All reactions were monitored by TLC (thin layer chromatography) on pre-coated Merk®silica gel 60F254plates. Column chromatography were performed by using silica gel Merck Geduran®Si 60 (40-63µm) of the crude product.
Synthesis Section
The synthesis and chemical analysis of imidazo[1,2-a]pyridine acrylonitriles bearing a piperazine group at position 6 (2b and 3c) has been reported in a previous work [5]. Here, other imidazo[1,2-a]pyridine acrylonitriles derivatives were reported.
General method for aldehydes synthesis (2a-b)
To a stirred solution of 2-aminopyridine (44.3 mmol, 1 eq) in DME, 100mL. After stirring at room temperature for 5h, the formed solid was filtered, and washed with thrice 25 mL of DME, 1,1,3-trichloroacetone (63.5 mmol, 1.4 eq) was added dropwise at room temperature. The off-ASA white solid was transferred to a second round-bottom flask. Then, ethanol (1L) was added, and the suspension was heated under reflux for 2h. After cooling, ethanol was evaporated, and the crude was alkalinized with 100mL of Na2CO3 aqueous saturated solution. The aqueous phase was extracted with 100mL of CH2Cl2 three times. The combined organic phases were dried over MgSO4 and evaporated to dry. The crude solid (8.3 g, 59%) of acetals (1a-b) was used directly for the next step without any purification.
To a stirred solution of acetals (e.g. 27 mmol, 1 eq) in CH3CN, 100 mL and H2O (33 mL), concentrated HCl (3 mL, 35.1 mmol, 1.3 eq) was added dropwise at room temperature. The reaction mixture was heated at 100°C for 3h. After cooling, the reaction mixture was alkalinized with Na2CO3 (ca. 3-4 g). The aqueous phase was extracted with thrice 60mL of CH2Cl2. The combined organic phases were dried over MgSO4 and evaporated to dry to afford 2a-b.
Imidazo[1,2-a]pyridine-2-carbaldéhyde (2a)
Black solid, 81%. 1H NMR (300MHz, CDCl3) δ: 10.20 (s, 1H, CHO), 8.32 (s, 1H, H-3), 8.31 (d, 1H, J = 6.7Hz, H-5), 7.82 (d, 1H, J = 9.3 Hz, H-8), 7. 41 (dd, 1H, J = 9.2, 6.8Hz, H-7), 7.02 (t, 1H, J = 6.8 Hz, H-6). 13C NMR (75MHz, CDCl3) δ: 188.1 (CHO), 143.8 (C-2), 126.8 (C-5), 126.7 (C-7), 119.5 (C-8), 115.6 (C-3), 114.6 (C-6). C-8a is missing. m.p.: 115 – 120 °C. HRMS (ESI): m/z calc. for C8H6N2O [M+H]+: 146.05383, found: 146.05321.
6-(4-Methylpiperazin-1-yl)imidazo[1,2-a]pyridine-2-carbaldehyde (2b) [5]
General method for Knoevenagel condensation
Method A: Method for acrylonitriles synthesis (3a-b)
To a stirred solution of MeONa in MeOH 5%, 1.5 mL, 2a (0.5 mmol, 1eq) was added, immediately followed by phenylacetonitrile (3.76 mmol, 1.1 eq) at room temperature. The reaction mixture was heated under reflux for 1h. After cooling for 1h in ice bath, the solid was filtered and washed with a minimum of cold MeOH. If not pure, crude product was purified by column chromatography on silica gel with CH2Cl2/MeOH: 99:1®90:10 as eluent.
Method B: Method for acrylamides (4) and acrylonitriles (8) synthesis
To a stirred solution of 2a (0.5 mmol, 1 eq) in EtOH, (1.5 mL), cyano derivative (1.1 eq) was added, followed by piperidine (10%, 5 µL). The reaction mixture was heated under reflux for 3h. After cooling for 1h in ice bath, the solid was filtered and washed with a minimum of cold MeOH. If not pure, crude product was purified by column chromatography on silica gel column, with a gradient of CH2Cl2® CH2Cl2 / AcOEt (80:20) as eluent.
(Z)-3-(Imidazo[1,2-a]pyridin-2-yl)-2-phenylacrylonitrile (3a)
Method A. Yellow solid, 30%. 1H NMR (300 MHz, CDCl3) δ: 8.55 (s, 1H, H-3), 8.15 (d, 1H, J = 6.8 Hz, H-5), 7.82 (s, 1H, C = CH), 7.69 ( d, 2H, J = 7.1 Hz, Ph-2,6), 7.60 (d, 1H, J = 9.2 Hz, H8), 7.42 (m, 3H, Ph-3,4,5), 7.22 (d, 1H , J = 8.3 Hz, H-7), 6.85 (t, 1H, J = 6.7 Hz, H-6). 13C NMR (75 MHz, CDCl3) δ: 144.9 (C-8a), 139.9 (C-2), 135.7 (C=CH), 133.5 (Ph-1), 129.4 (Ph-4), 129.2 (Ph-3,5), 126.2 (C-5), 126.1 (C-7), 125.8 (Ph-2,6), 118.4 (CN), 117.9 (C-8), 113.8 (C-6), 113.5 (C- 3), 111.6 (C=CH). m.p.: 157 – 161 ° C. HRMS (ESI): m / z calc. for C16H11N3 [M + H] +: 245.14834, found: 245.14739.
(Z)-2-(4-Chlorophenyl)-3-(imidazo[1,2-a]pyridin-2-yl)acrylonitrile (3b)
Method A. Yellow solid, 10%. 1H NMR (300 MHz, CDCl3) δ: 8.61 (s, 1H, H-3), 8.26 (dt, 1H, J = 6.8, 1.1 Hz, H-5), 7.98 (s, 1H, C = CH), 7.80 (d, 1H, J = 9.8 Hz, H-8), 7.67 (m, 2H, Cl-Ph-2.6), 7.45 (m, 3H, H-7 and Cl-Ph-3.5), 7.06 (td, 1H, J = 6.9, 1.0Hz, H-6).13C NMR (75 MHz, CDCl3) δ: 145.0 (C-2), 139.7 (C-8a), 136.0 (C = CH), 135.4 (Cl-Ph-4), 132.1 (Cl-Ph-1), 129.5 (Cl-Ph-3.5 and C-7), 127.1 (Cl-Ph-2.6), 126.2 (C-5), 118.1 (CN), 117.9 (C-8), 113.9 (C-6) , 113.7 (C-3), 110.4 (C = CH). m.p.: 183 – 187 °C. HRMS (ESI): m / z calc. for C16H11N3 [M + H] +: 279.06438, found: 279.06937.
(Z)-3-(6-(4-Methylpiperazin-1-yl)imidazo[1,2-a]pyridin-2-yl)-2-phenylacrylonitrile (3c) [5]
(2Z) -2-cyano-N-hexaneyl-3- (imidazo[1,2-a]pyridin-2-yl) prop-2-enamide (4)
Method B. Yellow solid, 82%. 1H NMR (300 MHz, CDCl3) δ: 8.44 (s, 1H, C = CH), 8.32 (s, 1H, H-3), 8.17 (d, 1H, J = 6.9 Hz, H-5), 7.72 ( d, 1H, J = 9.2 Hz, H-8), 7.32 (m, 1H, H-7), 6.92 (t, 1H, J = 7.1 Hz, H-6), 6.39 (sl, 1H, NH), 3.38 (m, 2H, CH2 n-Hexane), 1.60 (m, 2H, CH2 n-hexane), 1.34 (m, 6H, 3 CH2 n-hexane), 0.89 (m, 3H, CH3 n-hexane). 13C NMR (75 MHz, CDCl3) δ: 160.1 (CO), 145.6 (C-8a), 144.7 (C=CH), 138.2 (C-2), 127.0 (C-7), 126.2 (C-5), 118.6 (C-8), 117.3 (CN), 116.8 (C-3), 114.4 (C-6), 104.9 (C=CH), 40.7 (CH2 n-hexane), 31.6 (CH2 n-hexane), 29.5 (CH2 n-hexane), 26.7 (CH2 n-hexane), 22.6 (CH2 n-hexane), 14.1 (CH3 n-hexane). m.p.: 88 - 92 °C. HRMS (ESI): m / z calc. for C17H20N4O [M+H]+: 296.12947, found: 296.13129.
(Z) -3- (3-chloroimidazo [1,2-a] pyridin-2-yl) -2-phenylacrylonitrile (5)
To a solution of 100 mg of (imidazo [1,2-a] pyridin-2-yl)-phenylacrylonitrile (0.41 mmol, 1 eq) in 2 ml of acetonitrile, 93 mg (0.7 mmol; 1.7 eq) of NCS was added with stirring at room temperature. The reaction was left under stirring for 3 h. The precipitate formed was washed several times with water to remove all traces of succinimide and the product was obtained after drying under vacuum.
Yellow solid, 72%. 1H NMR (300 MHz, CDCl3) δ: 8.03 (m, 1H, H-5), 7.69 (m, 3H, H-8 and Ph-2,6), 7.56 (s, 1H, C=CH), 7.43 (m, 3H, Ph-3,4,5), 7.30 (m, 1H, H-7), 6.96 (m, 1H, H-6).13C NMR (75 MHz, CDCl3) δ: 144.3 (C-8a), 135.6 (C-2), 134.4 (Ph-1), 129.6 (Ph-4), 129.2 (Ph-3,5), 128.6 (C=CH), 126.5 (C-7), 126.3 (Ph-2,6), 122.9 (C-5), 118.7 (C-8), 117.6 (CN), 114.1 C-6), 113.8 (C=CH). A carbon (C-3) is missing. m.p.: 147-151 °C. HRMS (ESI): m/z calc. for C16H10ClN3 [M+H]+: 279.09283, found: 279.09204.
2-(chloromethyl) imidazo[1,2-a]pyridine (6)
To a solution of 2-aminopyridine (1 mmol, 1 eq) in 470 μL of 1,2-dimethoxyethane with stirring, 1,3-dichloroacetone (1.2 mmol, 1.2 eq) was added. The whole was left under stirring at room temperature for four hours. The 1,2-dimethoxyethane was evaporated off in vacuo and the solid was taken up in 1.1 mL of ethanol then the medium was heated at reflux overnight. After cooling the solution, the ethanol was evaporated off in vacuo. The solid was acidified with an NH4Cl solution and then neutralized with 1M NaOH. The obtained precipitate was filtered off and dried. It was pure and did not require any further processing.
White solid, 54%. 1H NMR (300 MHz, CDCl3) δ: 8.09 (d, 1H, J = 6.8 Hz, H-5), 7.61 (m, 2H, H-3 and H-8), 7.21 (m, 1H, H-7), 6.83 (t, 1H, J = 6.7Hz, H-6), 4.78 (s, 2H, CH2Cl). 13C NMR (75 MHz, CDCl3) δ: 126.0 (C-5), 125.7 (C-7), 117.7 (C-8), 113.1 (C-6), 111.0 (C-3), 39.5 (CH2Cl). C-2 and C-8a are missing. m.p.: 91 – 95 ° C. HRMS (ESI): m/z calc. for C8H7ClN2 [M+H]+: 166.08678, found: 166.09586.
Imidazo[1,2-a]pyridin-2-ylacetonitrile (7)
To a solution of 2-chloromethylimidazo[1,2-a]pyridine (1 mmol, 1 eq) in 2.5 mL of DMSO, 71.6 mg (1.1 mmol, 1.1 eq) of potassium cyanide was added. The whole solution was stirred at room temperature overnight. The product was extracted with chloroform (4x 5 mL) and dichloromethane (3 x 5 mL) and then washed with water (4x 10 mL) to remove the DMSO. The organic phases were combined and dried over magnesium sulfate and evaporated in vacuo. The solid obtained was purified by chromatography on a silica gel column, with a gradient of CH2Cl2® CH2Cl2 / MeOH (99: 1) as eluent.
Black pasty solid, 45%. 1H NMR (300 MHz, CDCl3) δ: 8.12 (m, 1H, H-5), 7.65 (s, 1H, H-3), 7.57 (d, 1H, J = 9.1 Hz, H-8), 7.23 (d, 1H, J = 8.7Hz, H-7), 6.85 (t, 1H, J = 6.8Hz, H-6), 3.95 (s, 2H, CH2CN). 13C NMR (75 MHz, CDCl3) δ: 135.17 (C-2), 127.2 (C-7), 126.3 (C-5), 116.8 (C-8), 114.0 (C-6), 110.7 (C-3), 18.0 (CH2CN). Two carbons (C-8a and CN) are missing. HRMS (ESI): m/z calc. for C9H7N3 [M+H]+: 157.18892, found: 157.19131.
(Z)-2-(Imidazo[1,2-a]pyridin-2-yl) -3-phenylacrylonitrile (8)
Method B. Yellow solid, 91%. 1H NMR (300 MHz, CDCl3) δ: 8.27 (s, 1H, C=CH), 8.14 (d, 1H, J = 8.9 Hz, H-5), 7.97 (dd, 2H, J = 6.6, 2.7 Hz, Ph-2,6), 7.89 (s, 1H, H-3), 7.64 (d, 1H, J = 9.1 Hz, H-8), 7.47 (m, 3H, Ph-3,4,5), 7.29 (m, 1H, H-7), 6.87 (td, 1H, J = 6.9, 1.0Hz, H-6). 13C NMR (75 MHz, CDCl3) δ: 145.8 (Ph-1), 142.0 (C=CH), 141.0 (C-2), 133.7 (C-8a), 130.8 (Ph-4), 129.6 (Ph-2 , 6), 129.1 (Ph-3,5), 126.7 (C-7), 126.2 (C-5), 117.8 (CN), 117.3 (C-8), 113.3 (C-6), 111.3 (C-3), 103.5 (C=CH). m.p .: 176 - 180 °C. HRMS (ESI): m / z calc. for C16H11N3 [M+H]+: 245.14445, found: 245.14407.
Biology
Antifungal screening by bioautography assay
Products in powder form were first solubilized in methanol for the preparation of stock solutions titrating to 1 mg/mL. From each of these stock solutions, a range of 10 dilutions of reason 2 was prepared. Then, 10 μL of each solution was deposited on glass plates in Silicagel 60 F254. The plates were previously developed in saturated tanks of a mobile chloroform-methanol water phase in a ratio (65:35:5) and then dried. In addition, Candida albicans fungal inoculum containing approximately 105 cells/mL was obtained by seeding three colonies of a pure strain for 24 to 48 hours in Tryptone Soya broth. This inoculum was spread on each chromatogram. The plates were incubated at 30°C after solidification of the agar for 24 hours. The plates obtained were then impregnated with an aqueous solution of methylthiazolyl chloride Tetrazolium and incubated for 2 to 4 hours. Areas of growth inhibition subsequently appeared as white spots on a purple background [12]. Only those products that showed an inhibitory zone at the 10 μg threshold amount were selected for the determination of Minimum Inhibitory Concentrations (MICs).
Determination of Minimum Inhibitory Concentrations (MIC) by microplate dilution technique
The evaluation of antifungal efficacy by determining the Minimum Inhibitory Concentrations (MICs) was made using the microplate dilution technique. This technique consists of putting in contact a Candida inoculum with an increasing dilution of selected products in 96 well microplates. The preparation of the fungal inoculum is done as previously described in the bioautography assay. The stock solutions of imidazo[1,2-a]pyridinyl phenylacrylonitrile were prepared with Dimethylsulfoxide (DMSO) at a concentration of 1 mg/ml and then diluted with BTS broth to obtain concentrated solutions at 100μg/mL. Subsequently, 100 μL of this dilution was deposited in the wells in the first column and 50 μL of BTS broth was distributed to the following wells. Subsequently, 50 μL was taken from the first 100 μL of the first well to achieve a range of dilutions increasing for reason 2. Finally, 50 μL of inoculum was distributed to the wells except for the last one, which served as a control to ensure that there was no contamination. The plates were incubated at 30 °C for 48 hours. For the revelation of the prepared microplates, 40 μL of aqueous solution of Methyl Thiazolyl Chloride Tetrazolium (MTT) at a concentration of 2.5 mg/mL was distributed to the wells and incubated for a further 30 minutes at room temperature. Wells containing still living cells turned yellow to purple as a result of mitochondrial dehydrogenase activity. The MIC was given by the lowest concentration at which MTT did not turn purple.
Result and Dissection
Chemistry
The synthesis of imidazo[1,2-a]pyridinyl phenylacrylonitrile derivatives and analogs started by the preparation of common imidazo[1,2-a]pyridin-2-carboxaldehyde intermediate 2a-b (Scheme 1).

Scheme 1: Preparation of imidazo[1,2-a]pyridine-2-carbaldehydes
First, the imidazo[1,2-a]pyridine ring formation between commercially available 2-aminopyridine and 1,1,3-trichloroacetone, at room temperature in DME and then in refluxing ethanol afforded acetals 1a-b in 59% yield. The acetals 1a-b were further converted into aldehydes 2a-b in 81% and 83% yield, respectively, by reaction with HCl. The imidazo[1,2-a]pyridinyl phenylacrylonitrile derivatives 3a-c and analogs 4 were synthesized following a Knoevenagel condensation reaction between imidazo[1,2-a]pyridin-2-carboxaldehyde and various phenylacetonitriles or 2-cyano-N-alkylacetamides (Scheme 2).
The synthesis of α-derivatives of imidazo[1,2-a]pyridinyl-acrylonitriles started with a condensation of 2 aminopyridine with 1,3-dichloroacetone in DME and then ethanolic medium. Then, cyanation of the formed chloromethylated compound was performed using potassium cyanide in dimethylsulfoxide (Scheme 3).

Scheme 2: Preparation of imidazo[1,2-a]pyridine-2-yl)acrylonitriles and acrylamides

Scheme 3: Preparation of imidazo[1,2-a]pyridine-2-yl)acetonitriles
Likewise, 8 was obtained by Knoevenagel condensation between imidazo[1,2-a]pyridin-2-acetonitrile and benzaldehyde (Scheme 4). All products were characterized by the usual spectroscopic methods.

Scheme 4: Preparation of imidazo[1,2-a]pyridine-2-yl)acrylonitrile (8)
Antifungal activity
The results obtained during the antifungal screening of imidazopyridinyl-arylacrylonitriles against C. albicans, C. tropicalis and C. glabrata strains by the bioautography method showed that 5 of our molecules were active on all three strains at the threshold quantity of 10µg, while the last compound presented an activity only against Candida albicans and Candida tropicalis.
In view of the above results, it is clear that these imidazo[1,2-a]pyridinyl-arylacrylonitriles which had previously exhibited anti-Haemonchus contortus activities, [5] have inhibitory properties against clinical strains of Candida, namely Candida tropicalis, Candida albicans and Candida glabrata. Indeed, the analysis of the results obtained established that:
The presence of the phenylacrylonitrile chain in position 2 of the imidazo[1,2-a]pyridine heterocycle leads to unsubstituted imidazo[1,2-a]pyridinyl-β-acrylonitrile (3a) having antifungal activity induced on the three strains of Candida with MICs ranging from 3.19 to 408 µM. In fact, with a MIC of 6.37 μM, 3a has a better activity compared with Kétoconazole (MIC = 94.1 μM) and Fluconazole (MIC = 326.5 μM) against Candida albicans.

Furthermore, these results reveal that the effectiveness of these acrylonitrile derivatives varies from one species of Candida to another, depending on the nature of the substituent present either on the benzene homocycle or on the imidazo[1,2-a]pyridine heterocycle.
Imidazo[1,2-a]pyridinyl-α-acrylonitrile 8, an isomer of position of 3a, did not improve antifungal activity against C. tropicalis and Candida glabrata with MICs of 3.2 µM and 408 µM, respectively. However, with a MIC of 408 µM, this derivative turned out to be at least 60 times less effective than its structural analogue (3a) against clinical strain of Candida albicans (MIC= 408µM). Thus, the displacement of nitrile from its β position (3a), to the α position (8) of the phenylacrylonitrile chain, decreased the efficacy against Candida albicans and led to the same activity of against C. tropicalis and Candida glabrata. These findings corroborated the importance of the isomeric position of nitrile on the acrylonitrile chain as it was observed in this series in antiparasitic activities [5].
Introducing a chlorine on para-position of the benzene homocycle (3b) did not enhance significantly the anticandidosis performance against Candida albicans (MIC= 178.7 µM) and Candida glabrata (MIC= 357.5 µM) compared with the non-chlorinated derivative (3a). In addition, this compound induced a significant improvement in the anticandidosis activities against Candida tropicalis with a MIC of 1.4 μM. Thus, such result confirms the importance of halogen atoms in the improvement of the antifungal activities as it was reported in series of antifungal azoles in therapeutic [6].
The N-methylpiperazine analogue of compound 3b (3c), with MICs ranging from 0.5 to 264.6 μM, retained its anticandidosis efficacy on the three strains with slight improvement in the activities. In addition, with MIC of 0.5 μM, 3c allowed the best anti-tropicalis activity of the series. This anticandidosis performance is even superior to that of Ketoconazole (MIC = 188.2 μM and Fluconazole (MIC = 0.6 μM) on this strain. This anticandidosis efficacy induced by the presence of methylpiperazine could be linked to a better solubility of compound 3c, since the methylpiperazine entity is responsible for the increase in water solubility in certain chemical series such as antibacterial quinolones [6].
Compound 5, with a chlorine atom in position 3 of imidazo[1,2-a]pyridine led to remarquable anticandidosis activities. In fact, the 3-chlorinated derivative with its MIC at 2.8 µM exhibited the best antifungal efficacy against the strain of Candida albicans. Such a performance twice as high as that of 3a confirms the importance of the presence of a substituent in position 3 of imidazo[1,2-a]pyridine, as in the case of Zolpidem already used in therapy as a psychotropic agent. In addition, 5 with MICs of 1.4 μM proved to be 2 times more effective than the unchlorinated derivative (3a, MIC = 3.2μM) against Candida tropicalis. But this modulation undertaken around the imidazo[1,2-a]pyridine ring did not make it possible to obtain the desired improvement in the anti-glabrata activities with a MIC of 357.5 μM.
Hexaneylamide derivative (4) for its part showed restricted efficacy on Candida strains. Indeed, the absence of the phenylacrylonitrile resulted in an annihilation of the anti-glabrata activities. Its activity was narrow to Candida albicans and Candida tropicalis with respective MICs of 5.3 and 1.3μM. The replacement of acrylonitrile by an acrylamide function led to an antifungal efficacy against C. albicans (MIC = 5.3 μM) similar to that of phenyl derivative (3a, MIC = 6.4μM). However, this derivative with MICs of 1,3 μM proved to be 2 times more effective against C tropicalis than the acrylonitrile derivative 3a (MIC = 3.2μM).
At the end of the antifungal screening, interesting trends in the structural activity relationship can be summarized as follows:
(i) Displacement of the nitrile to the β position did not improve anticandidosis activities;
(ii) para-chlorination of the benzene homocycle improved the activities against C. tropicalis in detriment of activities against C. albicans;
(iii) the replacement of the benzene homocycle by a hexaneylamine group led to excellent activity against C. albicans and C. tropicalis. However, this derivative has lost all activity against C. glabrata;
(iv) in contrary to C. albicans and C. tropicalis, the 3-chlorinated derivative (5) was found to be the most effective. In addition, this derivative exhibited the best efficacy regardless of the Candida species considered; and
(v) the best performance on C. glabrata was obtained with the piperazine derivative (3c).
Conclusion
This work is part of the development of new drug candidates to contribute to pharmacochemical research in the fight against fungal diseases. The pharmacochemical concepts used, have enabled us to obtain new imidazo[1,2-a]pyridinyl-arylacrylonitriles endowed with anti-Candida properties. The evaluation of the anticandidosis activities of imidazo[1,2-a]pyridinyl-arylacrylonitriles against three strains of Candida (C. albicans, C. glabrata, C. tropicalis) showed that:
- a) The best performance on Candida albicans was obtained with the chlorinated derivatives in position 3 of imidazo[1,2-a]pyridine (5) with a MIC of 8 µM;
- b) The doubly modulated with the chlorine in para position of the benzene homocycle and the methylpiperazine group in position 6 of imidazo[1,2-a]pyridine (3c) exhibited the best activity against Candida tropicalis with a MIC of 5µM. Besides, unlike Candida glabrata, with MIC of 264.6 µM, these compounds showed the best performance; and
- c) The best antimycotic profile on the three clinical strains of tested Candida was obtained with compound 5 with MICs varying between 1.4 and 5 µM.
Furthermore, the chemical modulations undertaken showed that:
- a) The displacement of nitrile from β position to α position of the phenylacrylonitrile chain did not improve the antifungal activity on the three strains of Candida;
- b) the enhancement of antifungal activities was subject to the introduction of a chlorine atom at position 3 of imidazo[1,2-a]pyridine; and
- c) the best anti-Candida non albicans profile was obtained with the double modulation possessing a chlorine atom in para position of the benzene homocycle and a methylpiperazine in position 6 of the imidazo[1,2-a]pyridine (3c).
Such results confirm that the pharmacochemical concepts used for the design of imidazo[1,2-a]pyridinyl-arylacrylonitriles remain relevant for the development of new biomolecules. These findings allow us to validate the imidazo[1,2-a]pyridinyl-arylacrylonitrile chemical profile as a potential new antimycotic pharmacophore.
Acknowledgement
The authors thank the ‘‘département d’analyses chimiques et S.R.M. bio-logique et médicale” (Tours, France) for chemical analyses. This work was supported by the SCAC department of embassy of France in Abidjan, Ivory Coast.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Authors' contributions
All authors contributed toward data analysis, drafting and revising the paper and agreed to be responsible for all the aspects of this work.
Conflict of Interest
We have no conflicts of interest to disclose.
HOW TO CITE THIS ARTICLE
Deto Ursul Jean-Paul N’Guessan , Songuigama Coulibaly, Fulgence Kondo Kassi, Pierre-Olivier Delaye, Mélanie Pénichon, Cécile Enguehard-Gueiffier, Hassan Allouchi, Mahama Ouattara. Synthesis and SAR of Imidazo[1,2-a] Pyridinyl-Phenylacrylonitrile Derivatives as Potent Anticandidosis Agents, J. Med. Chem. Sci., 2021, 4(6) 554-563
DOI: 10.26655/JMCHEMSCI.2021.6.3
URL: http://www.jmchemsci.com/article_137496.html
- Pfaller A., Diekema D., Clin. Microbiol. Rev., 2007, 20:133 [Crossref], [Google Scholar], [Publisher]
- Pappas G., Lionakis M.S., Arendrup M.C., Ostrosky-Zeichner L., Kullberg B.J., Nat. Rev. Dis. Primers., 2018, 4:18026 [Crossref], [Google Scholar], [Publisher]
- Kotey F.C., Dayie N.T., Tetteh-Uarcoo P.B., Donkor E.S., Dis. Res. Treat., 2021, 14:1 [Crossref], [Google Scholar], [Publisher]
- N'guessan D.U.J.P., Ouattara M., Coulibaly S., Kone M. W., Sissouma D., World J. Pharm. Res., 2018, 7:21 [PDF], [Google Scholar],
- N'Guessan J.D.U., Delaye P.O., Pénichon M., Charvet C.L., Neveu C., Ouattara M., Enguehard-Gueiffier C., Gueiffier A., Allouchi H., Med. Chem., 2017, 25:6695 [Crossref], [Google Scholar], [Publisher]
- Bryskier A., Antibiotiques: Agents antibactériens et antifongiques, Edition ellipses Paris, chapitre 43 :Antifongiques systémiques, 1999, 1216 [Google Scholar]
- Fleming F.F., Yao L., Ravikumar P.C., Funk L., Shook B.C.. Med. Chem., 2010, 53:7902 [Crossref], [Google Scholar], [Publisher]
- Uchida K., Nishiyama Y., Yamaguchi H., Infec. Chemother., 2004, 10:216 [Crossref], [Google Scholar], [Publisher]
- Meunier B., Comptes Rendus Chimie. 2011, 14:400 [Crossref], [Google Scholar], [Publisher]
)